
Abstract
Juvenile or type 1 diabetes (T1D) involves autoimmune-mediated destruction of insulin-producing β cells in the islets of Langerhans in the pancreas. Lack of insulin prevents the absorption and metabolism of glucose throughout the body by interfering with cell signaling. Cytokines have been shown to play a key role in β cell destruction and regulation of autoimmunity in T1D. The multiple roles of cytokines in T1D pathogenesis, regulation, and regeneration of β cells presents both promise and challenge for their use in immunotherapy. We found that mycobacterial adjuvants induce various regulatory T cells in the non-obese diabetic (NOD) mouse model of T1D. Cytokines produced by these cells not only regulate innate and adaptive immunity but also prevent the development of diabetes and partially restored normoglycemia in diabetic NOD mice. We discovered that adjuvant immunotherapy upregulated Regenerating (Reg) genes in the islets and induced interleukin 22 (IL-22)-producing Th17 cells. IL-22 is known to upregulate Reg gene expression in islets and could potentially induce regeneration of β cells and prevent their apoptosis. Therefore, cytokines both induce and regulate T1D and have the potential to regenerate and preserve insulin-producing β cells in the islets.
Introduction
Cytokines Play a Key Role in Autoimmunity in T1D
A variety of cytokines are found in the insulitis lesions both in humans and in animal models of T1D. These cytokines might promote T1D by causing β-cell destruction or may downregulate autoimmunity and suppress disease development. A number of cytokines have been shown to be important for the development of T1D at the level of the islets and systemically through activation of the autoimmune response (Rabinovitch 1993; Rabinovitch and Suarez-Pinzon 2007). The pathogenic mechanism of β-cell destruction is still not clear; classical T-cell effector pathways as well as many cytokines are dispensable in gene knockout and transgenic animal models (Rabinovitch and Suarez-Pinzon 2007). Proinflammatory cytokines produced by islet-infiltrating immune cells induce β-cell apoptosis in T1D. Cytokines such as interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), interferon α (IFN-α), IFN-γ, TNF-β, IL-2, IL-6, IL-12, IL-17, IL-18, IL-21, and IL-27 promote disease, whereas IL-4, IL-10, and transfoming growth factor β (TGF-β) are inhibitory (van Belle and others 2011). The cells involved in producing these cytokines include CD4+, CD8+, and natural killer (NK) T cells. Antigen-presenting cells (APC) such as dendritic cells and macrophages also contribute to this process. In this context, CD4+ Th1 cells characterized by their production of IL-2 and IFN-γ and Th17 cells producing IL-17 are pathogenic, whereas IL-4 and IL-10 producing Th2, IL-10 secreting Tr1 and Th17 (Nikoopour and others 2010), TGF-β secreting Th3, and IL-10, and TGF-β producing Treg cells are protective (Wraith and others 2004).
Combinations of some of the proinflammatory cytokines such as IL-1, IFN-γ, TNF-α, and TNF-β are highly cytotoxic to pancreatic islets, and they impair insulin secretion (Rabinovitch 1998). IFN-γ plays a dual role in destruction of β cells via the signal transducer and activator of transcription-1 (STAT-1) pathway and in protection via the interferon regulatory factor-1 (IRF-1) pathway (Gysemans and others 2008). IL-10 and TNF-α similarly have a dual role in T1D induction and prevention. Transgenic expression of IFN-α, IFN-γ, and IL-2 by β-cells in non-diabetes-prone mice induced β-cell destructive insulitis and autoimmune diabetes, whereas expression of TNF-α, TNF-β, and IL-6 induced insulitis that did not progress to β-cell destruction and diabetes (Rabinovitch 1998). In many autoimmune conditions both in experimental animal and in human disease, a major pathogenic role is ascribed to chronic inflammation causing cytokines produced by Th1 and Th17 cells. Th1 cytokines, particularly IFN-γ, also cause autoimmune activation by expanding the pathogenic T cell cells, and IFN-γ production is amplified by IL-18 (Boraschi and Dinarello 2006).
Administration of a number of cytokines has been shown to prevent diabetes development in non-obese diabetic (NOD) mice (Rabinovitch 1998). IL-2 prevents diabetes by correcting deficits in cytokine production (Serreze and Leiter 1988; Tang and others 2008; Goudy and others 2011) and IL-4 induces Th2 cells in the islets (Rapoport and others 1993). IL-2 may also be important for immunoregulation by the activation of CD4+ Treg cells (Grinberg-Bleyer and others 2010a). The effect of cytokines on autoimmune diabetes development depends on the dose, route, frequency, and time of administration in relation to disease development. Interestingly, we found that IFN-γ and TNF-α can also downregulate effector T cells by inducing apoptosis in diabetic NOD mice (Qin and others 2004) confirming previous studies (Campbell and others 1991). Treatment of prediabetic NOD mice with granulocyte-macrophage colony stimulation factor (GM-CSF) provided protection against T1D (Gaudreau and others 2007). Deletion of proinflammatory cytokines IL-1, IL-2, IL-6, TNF-α, TNF-β, and IFN-γ and by gene knockout delayed or reduced diabetes incidence (Rabinovitch and Suarez-Pinzon 2007). Deletions of IL-12 had no effect on disease, whereas IFN-γ deletion was found to delay but not prevent diabetes in NOD mice (Trembleau and others 2003). Blocking IL-1β activity with the IL-1 receptor antagonist is entering into clinical trials for several autoimmune diseases (Mandrup-Poulsen and others 2010; Dinarello 2011). These studies suggest that many cytokines participate in the autoimmune response that leads to β-cell destruction.
Cytokines Can Be Protective and Regulatory in T1D: Cytokine-Based Therapies for T1D
Treatment of autoimmune diseases with cytokines or anticytokine antibodies provides a systemic approach that offers both promise and challenges. This treatment represents a nonantigen-specific therapy that requires careful consideration for the timing, dose, and route of administration. For example, IL-18 and TNF-α accelerate or inhibit T1D in NOD mice depending upon the time of therapy. Cytokines such as IL-2 and IL-4 were shown to prevent induction of T1D in NOD mice (Rabinovitch 1998). The activation and proliferation of CD4+CD25+ Treg cells in NOD mice and in humans is related to IL-2. In NOD mice the insulin-dependent diabetes gene-3 (Idd3) is linked to the development of T1D. The Idd3 gene is associated with differential glycosylation of IL-2 (Podolin and others 2000). Secretion of IL-2 by peripheral blood cells is reduced in patients with T1D (Kaye and others 1986; Kukreja and others 2002). Recent studies suggest that low dose or inducible IL-2 therapy prevents and can reverse T1D by upregulating Treg cells in vivo (Grinberg-Bleyer and others 2010a; Goudy and others 2011).
The approaches used for blocking proinflammatory cytokines that promote T1D can be divided into 3 groups (Rabinovitch 1998). The first is blocking the generation and effector function of cytokines, the second is deletion or suppression of the cells producing the cytokines, and the third is the induction of Treg cells to block the effector function of cytokines. The blocking of cytokines directly has been explored by using antibodies to various inflammatory cytokines or their receptors, and by using soluble cytokine receptors or their receptor antagonists (Luo and others 2010). Immunosuppressive drugs that delete pathogenic cytokine-producing T cells, monoclonal antibodies against T cell receptor or costimulatory molecules, and immunotherapy with adjuvants, autoantigens, or their peptide epitopes have been used for immunotherapy of T1D. The use of anti-CD3 monoclonal antibody to the T cell receptor has proven highly efficacious both in NOD mice and in human diabetes (Chatenoud and others 1994; Herold and others 2002). Antibodies to costimulatory molecules on APC or T cells, including anti-B7, CTLA4-Ig, and anti-CD40L, hold promise to block effector T cells or to induce Treg cells (Boehm and Bluestone 2004). Using β-cell autoantigens such as insulin, glutamic acid decarboxylase-65, heat shock protein-65, or their peptides to induce tolerance or immunoregulation is a promising approach for antigen-specific immunotherapy (Eldor and others 2009; Luo and others 2010; Sabatos-Peyton and others 2010; van Belle and others 2011). Finally, agents that promote induction of Treg cells such as microbial agents (Singh and others 1998; Tian and others 2009; Nikoopour and others 2010) and α-galactosylceramide (van den Heuvel and others 2011) have shown promise in the treatment of T1D. Many of these treatments help in producing regulatory cytokines such as IL-4, IL-10, IL-35, and TGF-β. Treatments that induce deletion of autoreactive Th1-type cells and/or activation of Treg cells may be better then those that simply block the effector T cells or pathogenic cytokines, because they may induce long-lasting tolerance and may not require long-term immunosuppression (Rabinovitch 1998).
Complexity of Inflammatory Cytokine Circuits in T1D
Role of IFN-γ and TNF-α: paradox or regulatory circuit?
Th1 cells have for some time been considered to play a key role in inducing autoimmune diabetes. For example, established Th1 cell lines that express diabetogenic T cell receptors (eg, BDC2.5) have the ability to initiate disease in neonatal nonobese diabetic mice (Katz and others 1995). In addition, IL-12 administration accelerates T1D development in NOD mice, and the more rapid onset of disease correlates with increased Th1 cytokine production by islet-infiltrating cells (Trembleau and others 1995). However, NOD mice can also develop diabetes in the absence of the hallmark Th1 cytokine IFN-γ (Hultgren and others 1996) or the Th1 driving cytokine IL-12 (Trembleau and others 1999, 2003). These findings indicate that Th1 cytokines are not the sole contributing factor in the pathogenesis of diabetes. Disruption in expression of T-bet (Tbx21, a transcription factor-specific for Th1 cells) in the NOD mouse completely blocks insulitis and diabetes development (Esensten and others 2009). It has been shown that diabetogenic effector T cells can paradoxically help islet-specific Treg cells to provide sustained protection from diabetes (Grinberg-Bleyer and others 2010a). As this immunoregulatory circuit is disrupted in BDC2.5 Tbx−/− NOD mice, it is likely that the absence of Th1 effector T cells affects the number and suppressive function of Treg cells in the first instance, and diabetes in these TCR transgenic KO mice ultimately results from dysfunctional Treg cells, and not Th1 cells per se. Immunostimulation of NOD mice with mycobacterial adjuvant leads to increased levels of IFN-γ and IL-17 with protection from disease. This protective effect depends on IFN-γ as NOD mice deficient for this cytokine are resistant to the adjuvant therapy protocol (Serreze and others 2001). This is not restricted to the adaptive immune response as NK cells also are regarded as the cellular source of this increased IFN-γ (Lee and others 2008). The above feedback mechanism can explain the paradoxical finding that use of complete Freund's adjuvant (CFA) therapy raises inflammatory cytokines such as IFN-γ and IL-17 while still being protective. This treatment also expands CD4+CD25+Foxp+ T cells along inflammatory cytokines and establishes a new balance between regulatory and effector T cells with tolerogenic outcome (Tian and others 2009). We have shown the apoptosis of diabetogenic T cells in NOD mice by IFN-γ and TNF-α as a mechanism of protection after mycobacterial adjuvant therapy (Qin and others 2004). This raises the possibility that in certain circumstances, IFN-γ could have regulatory effect (Sobel and others 2002; Kim and others 2004; Jain and others 2008). Contrary to these studies, neutralization of IFN-γ (bray-Sachs and others 1991) or IL-17 (Emamaullee and others 2009) protects NOD mice from diabetes development. In general, effector cells with a type 1 cytokine profiles contribute to diabetes pathogenesis; however, Treg cells keep these effector T cells in check (You and others 2005; Feuerer and others 2009). In autoimmune diseases, effector T cells should be considered as a double-edged sword as a result of the bilateral relationship between Treg cells and effector T cells. A defect in this regulatory loop can tip the balance toward an autoimmune process.
Recent evidence for the role of TNF-α in expansion of Treg cells, as part of the complex interactive cell communication and cytokine network, led us to revisit our conceptual approach and interpretation of the overall role of this cytokine in autoimmunity. In an elegant study, activated islet-specific effector T cells (Teffs) were shown to strongly boost the expansion of islet-specific Treg cells, which acquired an activated phenotype at the site of Ag presentation (Grinberg-Bleyer and others 2010b). The cotransfer of preactivated effector T cells from BDC2.5 NOD mice with BDC2.5 Treg cells dramatically increased the proliferation of these Treg cells in the pancreas (Grinberg-Bleyer and others 2010b). Although IL-2 is indispensible for the survival of Treg cells, the increased Treg cell proliferation of boosted Treg cells was not due to IL-2 (Grinberg-Bleyer and others 2010b). TNF-α is considered to play a role in this protective phenomenon. Interaction of TNF-α with TNFR2 on Treg cells promoted expansion of Treg cells in vitro. Linking Treg cell expansion to effector T cell activation generates a feedback regulatory loop for dynamically tuning the size of Treg cell populations relative to their target Teffs locally.
Role of IL-2 and IL-21: antiparallel regulatory circuit
IL-2 and IL-21 production in NOD mice is under the control of Idd3, a putative susceptibility gene locus. A reduced level of IL-2 in NOD mice compared with NOD mice congenic for the B6 Idd3 locus (Idd3 B6) correlates with reduced function of Treg cells (Yamanouchi and others 2007; Sgouroudis and others 2008). These authors argue that the reduced levels of IL-2 in wild type (WT) NOD mice explain the reduced function of Treg cells in these animals (Yamanouchi and others 2007; Sgouroudis and others 2008). Relative to the NOD allele, the protective Idd3 B6 allele augments the amount of IL-2 and affords resistance to spontaneous and CD4+ T cell-induced T1D (Sgouroudis and others 2008). Contrary to these findings, others did not observe any difference in IL-2 mRNA or IL-2 protein production from NOD versus NODB6.Idd3 T cells (McGuire and others 2009). The ratio of Treg cells to effector T cells primarily in the pancreatic islets (rather than in the peripheral lymphoid organs) is the major factor in progression toward diabetes. IL-2 has a central role in keeping the balance in favor of the Treg cells in the inflamed islets (Tang and others 2008). In WT NOD mice there is enhanced apoptosis of Treg cells in the islets, and low-dose administration of IL-2 compensates for the defective survival of Treg cells in the islets through induction of CD25 and Bcl-2 expression (Tang and others 2008; Grinberg-Bleyer and others 2010a). The Idd3 B6 congenic NOD mice primarily favor T1D disease resistance in congenic NOD mice by increasing the cycling and function of Treg cells within the inflammatory environment of the pancreas (Sgouroudis and others 2008).
The Idd3 locus also contains the IL-21 gene, and this cytokine is considered necessary for the development of diabetes in the NOD (King and others 2004; Datta and Sarvetnick 2008; Spolski and others 2008; Sutherland and others 2009; McGuire and others 2011). Increased levels of IL-21 and its receptor have been observed in WT NOD mice (King and others 2004; McGuire and others 2009), and IL-21 mRNA levels increased in the pancreas as diabetes developed (Sutherland and others 2009). This increased responsiveness to IL-21 drives homeostatic expansion of immune cells in the otherwise CD4+ T cell lymphpenic NOD strain. In contrast, NOD mice congenic for Idd3 B6 do not show CD4+ T cell lymphopenia (King and others 2004). Also, mycobacterial adjuvant therapy of NOD mice increases T cell number and survival in protected animals. The increased levels of IL-21 and IL-21r in NOD mice have not been observed in Idd3B6 congenic NOD or CFA-treated NOD mice (King and others 2004). In general, comparison of Idd3 locus in B6 and NOD mice indicates high IL-2/low IL-21 conferred by the Idd3B6 protective allele versus low IL-2/high IL-21 in WT NOD. Deletion of IL-21 signaling leads to the abrogation of disease development (Spolski and others 2008). Ex vivo differentiation of Th17 cells is defective in NOD mice lacking IL-21R expression (Spolski and others 2008). Thus, it seems that the overall effect of the IL-2/IL-21 genes of NOD origin is to promote instability and apoptosis of T cells.
In comparison to Idd3B6 congenic NOD mice, the lymphpenia observed in NOD WT could be as a result of the imbalanced expression of both IL-2 and IL-21. Interestingly, significantly higher levels of expression of the regeneration genes, Reg2 and Reg3β (PAP), were found in the pancreas of Il21r-KO NOD mice. Thus, in the NOD both increased Reg gene expression and protection from the development of diabetes are associated with IL-21R deficiency (Spolski and others 2008). In line with the above findings, we have found an increase in Reg2 gene expression correlates with protection in NOD mice administered CFA (Huszarik and others 2010). In fact, we should consider the overall cytokine profile in the NOD pancreas since, in addition to their role in immunoregulation, a number of cytokines may also be involved in promoting regeneration of the islet β cells.
Neutralization of IL-21 in NOD mice reduces lymphocyte infiltration into islets and inhibits diabetes, and the absence of IL-21 signaling prevented islet allograft rejection (McGuire and others 2011). The diabetes resistance in IL-21R −/− NOD mice was associated with increases in IFN-γ and IL-17 (Sutherland and others 2009). Therefore, protection in IL-21R −/− NOD mice is not because of reduced IL-17 production. More specifically, IL-21 neutralization leads to a sustained reduction in CD8+ T-cells with an activated/memory-phenotype, and prevents islet graft rejection (McGuire and others 2011). As CFA reduces IL-21 in NOD mice (King and others 2004), it would not be surprising that immunotherapy with mycobacterial adjuvant prevents recurrence of diabetes in syngeneic grafted islets (Wang and others 1992).
In conclusion, the common theme in the discrepant results of the studies mentioned above is that the Idd3 locus in NOD mice contributes to low IL-2 and high IL-21 cytokine profiles. Lower levels of IL-2 followed by decreased Teg cells in the islets might boost IL-21 dependent expansion of diabetogenic T cells. Similar to what we discussed above regarding TNF-α, IL-21 is another cytokine that exerts its regulatory effects at the interface between effector and Treg cells. A central idea here is that the effector T cells are no longer responsive to Treg cell suppression in mice with elevated levels of IL-21 (Clough and others 2008). Autoimmunity might be considered to result from resistance of the effector T cells to Treg cell-mediated suppression (You and others 2005). Early in life CD4+ CD25+ Treg cell are able to prevent diabetes; however, effector T cells acquire resistance to Treg cell suppression during progression toward diabetes and ultimately overt disease develops. IL-21 acts on effector T cells rather than Treg cells since Treg cells derived from high IL-21-expressing NOD diabetic animals retained the capacity to suppress effector cells (You and others 2005).
Role of IL-17 in T1D: duality of function
It has been shown that Th17-polarized BDC2.5 T cells can induce diabetes in severe combined immune deficient (SCID)/NOD mice (Bending and others 2009). However, instability of these cells in vivo and conversion to a Th1 phenotype precluded the conclusion that Th17 cells are directly involved in diabetes pathogenesis (Bending and others 2009). In contrast, other authors have found that adoptive transfer of Th17-polarized BDC2.5 cells made by another method (and thought to be more stable in NOD mice) induced pancreatitis but not diabetes (Martin-Orozco and others 2009). This could possibly be due to polarization of these cells with TGF-β and IL-6. Studies in other autoimmune models such as the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis have shown dual function for Th17 cells. Polarization with IL-23, IL-1β, and IL-6 induced pathogenic Th17 cells (Ghoreschi and others 2010), whereas TGF-β, IL-1β, and IL-6-polarized cells are not able to induce disease (McGeachy and others 2007). We have shown that in NOD mice, polarization with IL-23 plus IL-6 can induce disease and IL-1β is not required for Th17 polarization (unpublished data). Th17 cells that have been matured with TGF-β and IL-6 have high expression of the genes encoding RORγt, IL-17A, IL-17F, and IL-21 and also produce IL-10 and have been shown to be capable of regulatory function (McGeachy and others 2007). We have shown that polarized Th17 cells from CFA or Bacillus Calmette-Guerin (BCG) immunized NOD mice prevented adoptive transfer of disease (Nikoopour and others 2010). These studies indicate that the regulation of Th17 differentiation is potentially effective for the immunoregulation of autoimmune diseases. Due to the plasticity of these cells (Nurieva and others 2009), experiments in immune-competent rather than immune-deficient animals might resolve the discrepancy. As TGF-β+IL-6-polarized Th17 cells failed to induce disease in NOD mice, polarization with the more pathogenic cytokine conditions such as IL-23 plus IL-6 could lead to the diabetes development in NOD mice. To summarize this section, we have shown that BDC2.5 CD4+ T cells preactivated in the presence of IL-23 plus IL-6 are diabetogenic upon adoptive transfer into NOD mice, whereas TGF-β plus IL-6-polarized cells are regulatory (our unpublished data).
IL-10: opposing functions in contraction and expansion of Treg cell populations
There are discrepant findings on the role of IL-10 in autoimmune diabetes. Many studies have shown an association between increased IL-10 production and attenuation of autoimmune diabetes, suggesting that IL-10 has a protective effect (Goudy and others 2001, 2003). Contrary to those studies, transplantation of islets overexpressing IL-10 via adenoviral gene transfer failed to suppress rejection of the syngeneic grafts (Smith and others 1997). Also, local expression of IL-10 in islets accelerated diabetes (Lee and others 1996). The absence of endogenous IL-10 in NOD.IL-10−/− mice did not accelerates diabetes in a specific pathogen-free, clean environment (Serreze and others 2001). However, NOD.IL-10−/− mice raised in the conventional facility showed reduced development of spontaneous T1D, suggesting that exposure to normal environmental microflora almost completely protected NOD.IL-10−/− mice from spontaneous diabetes (Rajagopalan and others 2006). It seems that the major regulatory pathway controlling autoimmunity is Treg cells, and IL-10 does not have a pivotal role in protecting NOD mice from diabetes. Diabetes in NOD mice could be prevented by immunization with CFA, and this effect has been shown to be independent of endogenous IL-10 in NOD mice (Serreze and others 2001).
CD4−CD8− “double negative” (DN) T cells, specific for a pancreatic neo-antigen, have suppressive activity through specific lysis of B cells loaded with neo-self antigens (Hillhouse and others 2010). NOD mice have a reduced number of DN T cells, and passive transfer of DN T cells prevents diabetes progression (Dugas and others 2010). Exposure of DN T cells to IL-10 leads to apoptosis. This might explain the discrepant results regarding IL-10's role in NOD mice. While IL-10 acts on Foxp3+ Treg cells to enhance Foxp3 and IL-10 expression, it also acts on DN T cells to enhance apoptosis, thus resulting in a reduction of this regulatory population. These results suggest that IL-10 is part of the cytokine regulatory network, and the balance between these 2 different populations of Treg cells needs to be considered to discern the overall effect of IL-10 on protection versus exacerbation of autoimmune diabetes.
Cytokines and Islet β Cell Regeneration
Cytokines are able to induce differentiation of various lymphoid and nonlymphoid cells. Adjuvants provide a powerful stimulus to induce cytokine production and could regulate various innate and adaptive immune responses. We found that a single injection of the mycobacterial adjuvants, CFA or BCG, prevented the development of diabetes in NOD mice by downregulating autoimmunity (Sadelain and others 1990; Qin and others 1993). CFA treatment also restored normoglycemia in a small percentage of end-stage diabetic NOD mice (Kodama and others 2003; Chong and others 2006; Faustman and others 2006; Nishio and others 2006; Suri and others 2006). This was evident by the reappearance of pancreatic β-cells as observed by histological analysis of the islet tissue (Qin and others 1993; Huszarik and others 2010).
Reg genes and islet β cell regeneration and protection
The role that mycobacterial adjuvants such as BCG and CFA play in the regeneration of β-cell mass is unclear. Prevention of autoimmunity by itself is not enough to promote β-cell regeneration in diabetic mice, as immunotherapy using anti-CD3 monoclonal antibody was not found to be accompanied by β-cell regeneration (Ablamunits and others 2007; Phillips and others 2007). Several years ago inflammatory stimuli were suggested to induce upregulation of the Regenerating (Reg) family of genes within the islets (Okamoto and Takasawa 2002). These proteins are secreted and can act through their cognate receptors to stimulate β-cell replication in an autocrine and/or paracrine manner. This led us to hypothesize that CFA treatment could stimulate β-cell regeneration in diabetic NOD mice not only through prevention of autoimmunity, but also by upregulating Reg genes and preventing their apoptosis (Fig. 1). A number of Reg genes as islet-regenerating growth factors could be involved in endogenous β-cell regeneration, such as Reg1, Reg2, and Reg3β. The Reg protein family has been implicated in islet β-cell proliferation, survival, and regeneration and is a part of a larger group of calcium-dependent carbohydrate binding proteins (C-type lectins) (Liu and others 2008). These small, secreted proteins act as trophic and/or antiapoptotic factors and/or survival/growth factors for insulin-producing pancreatic β-cells, neural cells, and epithelial cells of the digestive system. They also have a role in tumor formation and are potential biomarkers of carcinogenesis. Reg2 is one such molecule and may function as a growth factor that enhances the survival and function of islet cells. Mouse Reg2 is expressed in regenerating islets and in the normal exocrine pancreas. Reg2 also stimulates the growth of pancreatic β cells (Baeza and others 1997; Planas and others 2006; Gurr and others 2007).
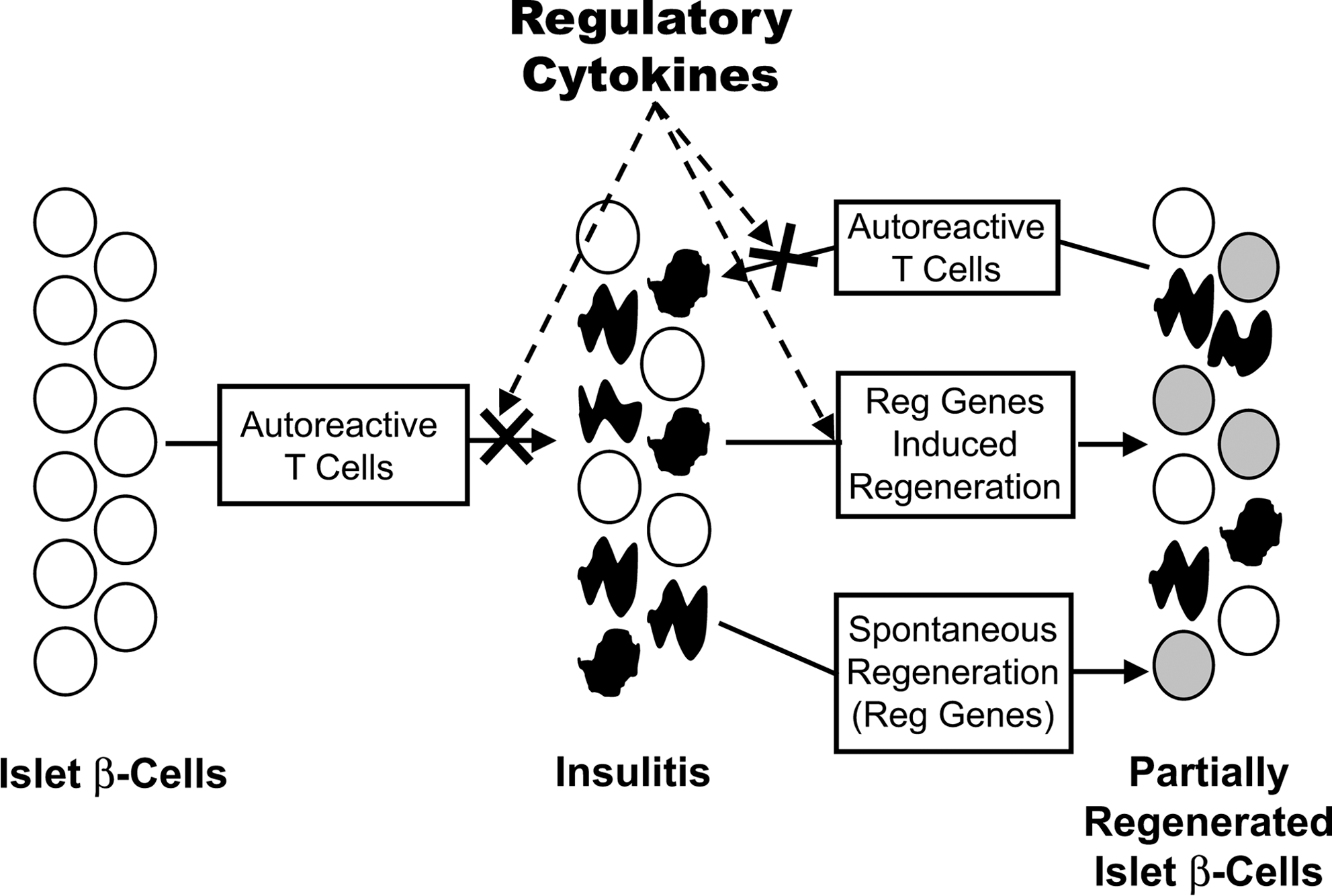
Islet β-cell cell regeneration after blocking of autoreactive T cells. Insulitis leads to the destruction of β-cells, which stimulates a regenerative response mediated by the Reg genes. This allows regeneration of some β-cell mass to occur; however, the ongoing autoimmunity destroys the newly regenerated cells and diabetes eventually ensues. Adjuvant immunotherapy of diabetic NOD mice blocks autoimmunity and allows the upregulation of Reg genes, which results in regeneration and preservation of β-cell mass and partial reversal of diabetes.
We recently showed that CFA treatment does indeed cause a significant upregulation of Reg2 expression in both diabetic and nondiabetic animals (Huszarik and others 2010). This effect is not dependent on signalling through MyD88 or through IL-6 that was previously linked to the regulation of Reg gene expression (Planas and others 2006). We also found Reg2 is upregulated in mice that were treated with β-cell regenerative islet neogenesis associated protein (INGAP) peptide or with Exendin-4. The Reg2 expression correlated with a partial reversal of insulitis and an increase in insulin production. Upon CFA treatment we also found improved glucose tolerance tests in streptozotocin (STZ)-treated diabetic C57BL/6 mice and an increase in the number of small islets in the pancreas of diabetic NOD mice. Therefore, immunotherapy with adjuvants induces Reg2-mediated partial regeneration of β-cells (Huszarik and others 2010).
In a recent study Liu and others (2010) reported that pancreatic islet-specific overexpression of Reg3β protected mice against STZ-induced diabetes (Xiong and others 2011). Reg2 provided protection of insulinoma cells from STZ-induced mitochondrial damage but no protection of STZ-induced diabetes by transgenic expression of Reg2 in the acinar cells (Li and others 2010). The level of Reg2 protein expression within the pancreas in these transgenic mice led to a moderate 2.8-fold increase in protein expression. This sub-optimal level of Reg2 protein expression has implications for the conclusions drawn by the authors. The protein expression is patchy and located in the exocrine pancreas only. The choice of acinar cells for the location of transgenic Reg2 expression does not address the role of Reg2 in diabetes and β cells. We found that CFA treatment induced a 60- to 200-fold increase in Reg2 mRNA levels in both B6 and prediabetic NOD mice and a large increase in Reg2 protein expression by western blot. The Reg2 was localized throughout the islets and most strongly in the β cell areas, and there was very little expression in the exocrine pancreas (Gurr and others 2007).
Mechanism for adjuvant-mediated upregulation of Reg genes and β cell regeneration in the pancreas
We propose that cytokines induced by adjuvants act on islet cells to activate transcription factors that lead to the production of Reg family of growth factors such as Reg1, Reg2, and Reg3β (PAP1) (Fig. 2). These growth factors act in an autocrine fashion to induce β cell differentiation. We found that immunization with CFA induced Th17 cells that produced IL-17, IL-22, IL-10, and IFN-γ in NOD mice (Nikoopour and others 2010). IL-22 is known to upregulate Reg gene expression in islets and could potentially induce regeneration of β cells. It has been shown that the highest level of expression of IL-22 receptor (IL-22R) mRNA is in the pancreas (Aggarwal and others 2001; Gurney 2004). In the human pancreas, IL-22 receptor is expressed in the α and β cells and not by the acinar cells or ductal epithelium (Shioya and others 2008). Pancreatic cells respond to IL-22 with the activation of Stat3 and upregulation of various Reg genes both in mouse and in human cells (Aggarwal and others 2001; Gurney 2004; Sekikawa and others 2010; Wolk and others 2010). Reg proteins are known to induce islet growth and differentiation and prevent their apoptosis (Baeza and others 1997; Liu and others 2010; Xiong and others 2011). In vivo injection of IL-22 resulted in rapid induction of mRNA for Reg gene family members such as Reg3β (PAP1) in pancreas (Aggarwal and others 2001). These results support the conclusion that CFA-mediated upregulation of IL-22 leads to the induction of Reg genes in pancreas and the Reg proteins in turn induce the differentiation of β cells in the pancreas and prevent their apoptosis (Fig. 2).

Cytokine-mediated upregulation of transcription factors and Reg proteins. Cytokines induced by adjuvant immunotherapy activate transcription factors in pancreatic tissue that stimulate production of Reg proteins. These secreted Reg proteins induce islet growth and differentiation and prevent β-cell apoptosis. STAT, signal transducer and activator of transcription; TFs, transcription factors.
Conclusions
Over the past 20 years considerable data have emerged from studies in animal models and in human diabetes to illustrate that cytokines play a key role in β cell destruction and regulation of autoimmunity involved in T1D. The dual role of cytokines expressed in islets in the pathogenesis and regulation of T1D presents considerable challenges for the therapy of both type 1 and type 2 diabetes. Cytokines are likely to be involved in the normal turnover and regeneration of β cells in islets. Moreover, β cell mass is dynamic and capable of undergoing adaptive changes in response to differing requirements for insulin such as occur in pregnancy, obesity, and diabetes. β cell replication continues over a lifetime at a low pace and two pathways have been suggested as a source of new β cells for this process. One is based upon the replication of pre-existing β cells in the pancreas, and the other proposes that new β-cells may arise from stem cells residing in the ductal epithelium (Bonner-Weir and others 2010).
Our work suggests that cytokines play a key role in regulating β cell destruction and regeneration. We found that the Reg gene family is upregulated during diabetes development in a situation where the gene products are likely to stimulate β-cell regeneration. However, this regenerative response cannot compensate for β-cell loss due to autoimmune destruction. Experimental induction of cytokines such as IL-22 by adjuvants can upregulate the expression of Reg genes, which could potentially induce regeneration of β-cells in islets (Gurney 2004; Wolk and others 2010). Our studies suggest that Regenerating (Reg) gene family members such as Reg2 can be induced by cytokines. This provides a useful target for β-cell regenerative therapies (Gross and others 1998; Dungan and others 2009).
Footnotes
Acknowledgments
The authors wish to acknowledge Edwin Lee-Chan for editorial assistance in the preparation of the article. This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC). A. M. Jevnikar is supported by a Wyeth-CIHR Clinical Chair in Transplantation.
Author Disclosure Statement
No competing financial interests exist.