
Abstract
Interleukin-18 (IL-18) is a highly regulated inflammatory cytokine that is elevated in synovial tissues and synovial fluids of patients with rheumatoid arthritis (RA) compared with patients with osteoarthritis (OA) and patients with other arthropathies. Within the RA joint, IL-18 can contribute to the inflammatory process by inducing leukocyte extravasation through upregulation of endothelial cell adhesion molecules, the release of chemokines from RA synovial fibroblasts, and directly as a monocytes, lymphocyte, and neutrophil chemoattractant. IL-18 can also help maintain and develop the inflammatory pannus by inducing endothelial cell migration and angiogenesis. IL-18 does this directly by binding and activating endothelial cells and indirectly by inducing RA synovial fibroblasts to produce angiogenic chemokines and vascular endothelial growth factor. IL-18 is present in RA synovial fluid in high levels, where it functions as an angiogenic mediator and leukocyte chemoattractant. IL-18 mediates all these inflammatory processes by binding to its receptor, IL-18 receptor, and initiating the activation of different signaling cascades leading to changes in target cells gene expression and behavior. IL-18 has been identified as a potential therapeutic target in the treatment of RA.
Interleukin-18
IL-18 Binding Protein
IL-18 binding protein (IL-18BP) is a constitutively expressed high affinity receptor for IL-18 (Novick and others 1999). There are 4 different human isoforms of IL-18BP(a-d) created by alternate splicing with isoforms a and c capable of binding IL-18 (Kim and others 2000). IL-18BPa is the major splice variant and has the strongest binding affinity of the isoforms. The primary activator of IL-18BP production is IFN-γ and as such creates a negative feedback loop controlling IL-18 levels in which IL-18-induced IFN-γ results in increased IL-18BP and reduced IL-18 levels (Moller and others 2003).
IL-18 Regulation
IL-18 biologic activity is highly regulated by several different mechanisms. First, it must be expressed, and then it has to be cleaved releasing the biologically active portion. Caspase-1, the primary enzyme responsible for pro-IL-18 cleavage to its active mature form, is also highly regulated. However, other non-caspase-1 mechanisms can generate biologically active IL-18, including proteinase 3 cleavage (Sugawara and others 2001). Once released, IL-18 can be bound by either IL-18BP or soluble IL-18 receptor (IL-18R), preventing its biologic activity. If active IL-18 circumvents these regulatory mechanisms, its activity is still mediated by other cytokines with which it functions in synergy, such as IL-12 and IL-15 (Yamamura and others 2001; Gracie and others 2003). Additionally, expression of the IL-18R determines the cell type that will respond to IL-18. Finally, the signaling pathways will determine the activation of the cells both producing the IL-18 and being stimulated by IL-18. Ultimately, the biologic activity of IL-18 is not only determined by the production of IL-18 but also the production of other molecules, including caspase-1, IL-18BP, soluble IL-18R, IL-12, IL-15, IL-18R, and several signaling cascades.
IL-18's Role in Rheumatoid Arthritis
In rheumatoid arthritis (RA), IL-18 is significantly elevated in sera, synovial tissues, and synovial fluid compared with those from osteoarthritic patients and patients with other arthropathies (Gracie and others 1999; Yamamura and others 2001; Bresnihan and others 2002). IL-18 levels correlate with acute phase proteins and disease activity (Yamamura and others 2001; Petrovic-Rackov and Pejnovic 2006; Shao and others 2009). In the RA joint, IL-18 is primarily produced by synovial lining macrophages; however, it is also produced by synovial fibroblasts, endothelial cells, dendritic cells, articular chondrocytes, osteoblasts, and synovial fluid neutrophils (Gracie and others 1999; Moller and others 2002; Verri and others 2007). In vitro, IL-18 is expressed by CD68+ macrophages, neutrophils, and RA synovial fibroblasts. Additionally, IL-18 expression can be induced by cytokine stimulation of RA synovial fibroblasts and RA peripheral blood neutrophils (Gracie and others 1999; Verri and others 2007). While there have been reports that RA synovial fibroblasts produce IL-18 mRNA but do not make mature biologically active IL-18 (Yamamura and others 2001; Zeisel and others 2004), we recently reported that RA synovial fibroblasts release biologically active IL-18 (Marotte and others 2010). Specifically, tumor necrosis factor-α (TNF-α) stimulation of RA synovial fibroblasts results in IL-18 expression and production of biologically active IL-18. This activation of RA synovial fibroblasts by TNF-α is dependent upon the activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway. Based upon their production in the RA joint, there is potentially a positive feedback loop between IL-18-producing synovial fibroblasts and TNF-α-producing, synovial macrophages, which could result in the escalation of inflammation seen in RA.
IL-18 stimulates synovial lymphocytes, macrophages, and fibroblasts through binding to the IL-18Rs on the surface of these cells (Gracie and others 2003). While there is some contention as to whether synovial fibroblasts produce both α and β chains of the IL-18R (Kawashima and Miossec 2003) and whether they respond to IL-18 stimulation (Moller and others 2002), we and others have shown RA synovial fibroblasts to respond directly to IL-18 stimulation resulting in the activation of signaling cascades and the initiation of the expression of cytokines, adhesion molecules, and angiogenic mediators (Morel and others 2001a, 2001b, 2002; Ruth and others 2003; Cho and others 2006; Amin and others 2007). Specifically, in response to IL-18 in vitro, RA synovial fibroblasts increase their production of adhesion molecules, intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), chemokines, IL-8/CXCL8, epithelial neutrophil-activating peptide 78 (ENA-78/CXCL5), growth-regulated oncogene-alpha (GROα/CXCL1), stromal cell-derived factor-1 alpha (SDF-1α/CXCL12), monocyte chemotactic protein-1 (MCP-1/CCL2), and macrophage inflammatory protein-3 alpha (MIP-3α/CCL20), and an angiogenic mediator vascular endothelial growth factor (VEGF), all of which are integrally involved in the pathogenesis of RA. Thus, IL-18 stimulates synovial fibroblasts to release mediators resulting in leukocyte recruitment and activation as well as new blood vessel formation. Additionally, IL-18 acts in synergy with IL-12 to stimulate T cell production of IFN-γ, which in turn stimulates synovial macrophages to produce TNF-α, leading to joint inflammation, and IL-1β, resulting in cartilage destruction (Fig. 1).
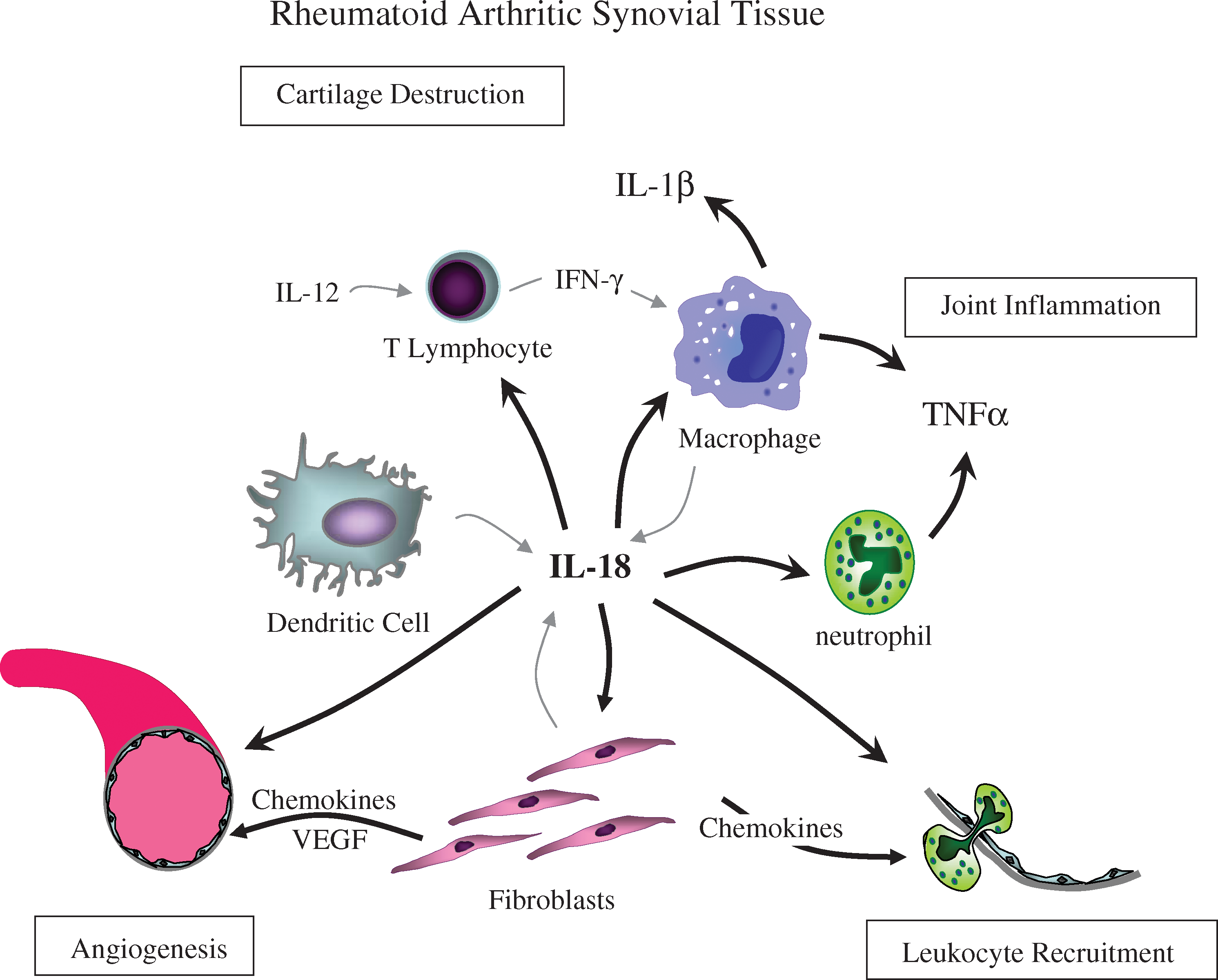
IL-18 mediates joint inflammation, cartilage destruction, angiogenesis, and leukocyte recruitment in rheumatoid arthritis. IL-18, interleukin-18; IL-12, interleukin-12; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; IL-1β, interleukin-1β; VEGF, vascular endothelial growth factor.
Role of IL-18 in Pathogenesis of Animal Models of Arthritis
In animal models of arthritis, IL-18 increases inflammation and cartilage erosion, whereas treatment with IL-18BPa or anti-IL-18 reduces arthritis, joint erosion, and inflammatory cytokine expression (Haas and others 2006). Specifically, in studies using streptococcal cell wall-induced arthritis in mice, intra-articular injections of anti-IL-18 antibody significantly reduced joint swelling and TNF-α and IL-1 levels (Joosten and others 2000). This reduction in inflammation and arthritis was independent of IFN-γ, as IFN-γ gene-deficient animals with arthritis also responded to the anti-IL-18 treatment. IL-18 administration to collagen-induced arthritis (CIA) mice resulted in heightened arthritis and cartilage erosion (Gracie and others 1999). IL-18 gene-deficient mice also have reduced arthritis in various arthritis models, validating a role for IL-18 in the pathogenesis of RA (Wei and others 2001). For example, in the zymosan-induced arthritis model, IL-18 gene-deficient mice, in addition to reduced arthritis, showed a reduction in the monocyte chemoattractant MIP-3α/CCL20, angiogenic mediator VEGF, and IL-17 (Ruth and others 2010). IL-18 was shown to induce joint inflammation independent of IL-1 and cartilage destruction independent of TNF-α using IL-1 or TNF-α gene-deficient mice (Joosten and others 2004). These models suggest a central role for IL-18 in mediating the joint inflammation through initiating inflammatory cytokine production, leukocyte extravasation, angiogenesis, and the Th17 autoimmune response.
Synovial Fibroblast Response to IL-18 Stimulation
Central to the role of IL-18 in the pathogenesis of RA is the response of the synovial fibroblast to IL-18 stimulation. In vitro studies by our group and others have shown that IL-18, at concentrations found in RA synovial fluids, can induce the production of neutrophil chemoattractants, IL-8/CXCL8, ENA-78/CXCL5, and GROα/CXCL1 (Morel and others 2001a). This induction of IL-8/CXCL8 was direct, not requiring stimulation with other cytokines such as IL-12, and involved signaling through nuclear factor kappa B (NFkB). Additionally, IL-18-stimulated RA synovial fibroblasts express increased levels of adhesion molecules ICAM-1 and VCAM-1 (Morel and others 2001b). This elevation in adhesion molecule expression by synovial fibroblasts involved IL-18 activation of multiple signaling pathways (Morel and others 2002). IL-18 induction of RA synovial fibroblast VCAM-1 involved activation of IRAK/NFkB pathway and Src kinase activation of both Ras/Raf/ERK/activator protein-1 (AP-1), and phosphatidylinositol-3-kinase (PI3K)/Akt pathways (Fig. 2).
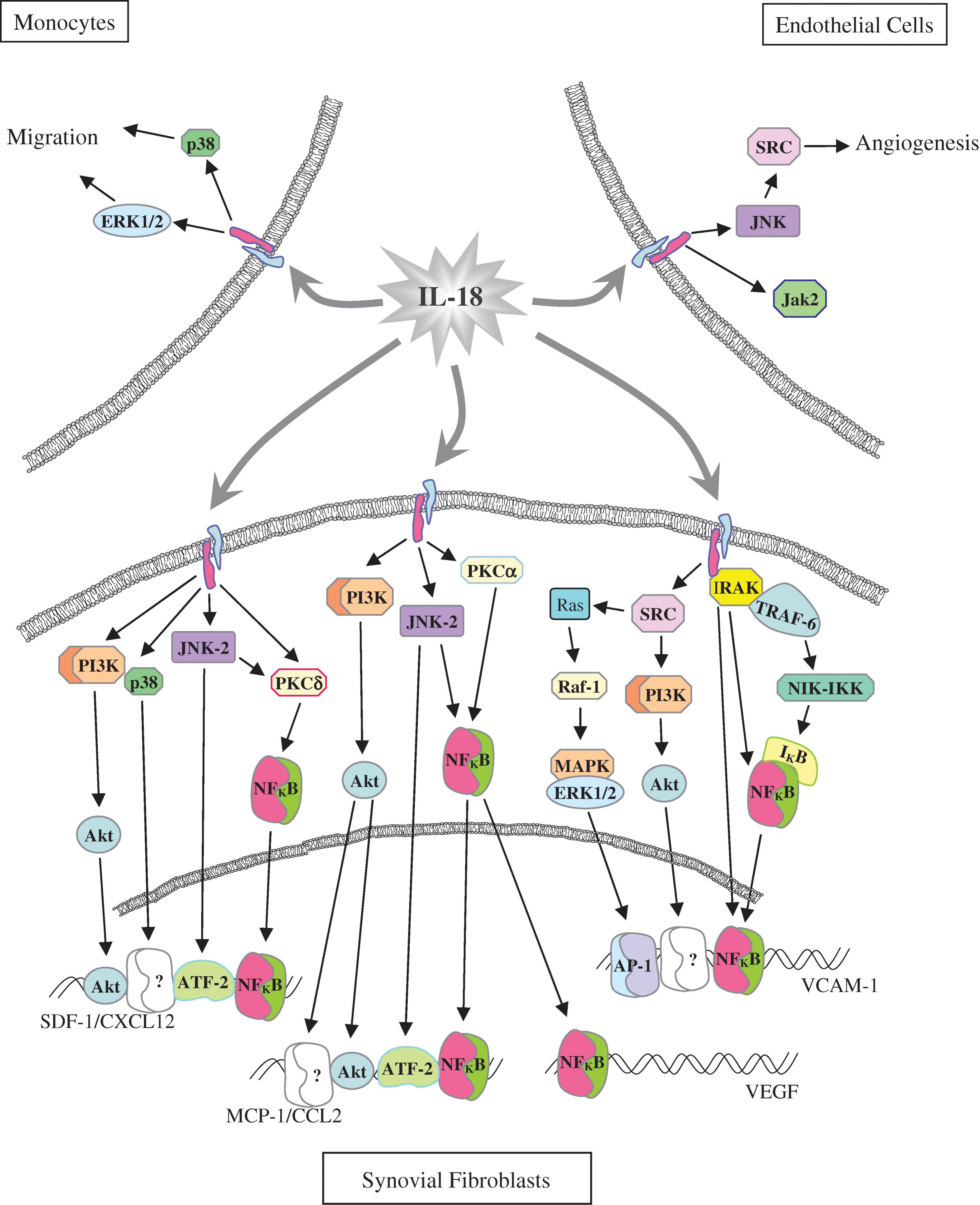
IL-18 initiated signaling pathways in synovial fibroblasts, monocytes, and endothelial cells. ATF-2, activating transcription factor-2; AP-1, activator protein-1; JNK, c-Jun N terminal kinase; ERK1/2, extracellular signal-regulated kinase 1/2; IKB, inhibitory kappa B (KB); IKK, IKB kinase; IRAK, IL-1 receptor-associated kinase; Jak2, janus kinase; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor kappa B; NIK, NFκB-inducing kinase; PI3K, phosphatidylinositol-3-kinase; PKC, protein kinase C; Src, Src kinase; TRAF-6, TNF receptor-associated factor-6.
IL-18 Mediates Joint Inflammation
IL-18 also mediates joint inflammation by augmenting leukocyte extravasation through induction of endothelial cell adhesion molecule expression. In vitro experiments using human microvascular endothelial cells (HMVECs) showed that IL-18 stimulation could induce adhesion molecule expression (Morel and others 2001b). Specifically, IL-18 activated both NFkB and PI3K signaling pathways resulting in endothelial cell expression of E-selectin, involved in leukocyte rolling, and ICAM-1 and VCAM-1 involve in leukocyte firm adhesion. This was verified in experiments showing that IL-18 treatment of endothelial cells resulted in increased adhesion of HL-60 cells which was dependent upon leukocyte ligands for E-selectin and ICAM-1 and VCAM-1.
IL-18 is a chemoattractant for synovial CD4+ T lymphocytes from patients with RA (Komai-Koma and others 2003). IL-18 is also important in RA peripheral blood and synovial fluid neutrophil migration and activation. IL-18R is constitutively expressed on neutrophils and RA synovial fluid neutrophils stimulated with IL-18 in vitro produce greater amounts of chemokines and cytokines (Leung and others 2001). IL-18-induced neutrophil migration is dependent upon downstream cytokines (TNF-α and Leukotriene B4 [LTB4]) and chemokine (MIP-2/CXCL2 and MIP-1α/CCL3) production. This was validated in the mouse CIA model, in which arthritis was enhanced by IL-18 in part through increased neutrophil recruitment involving TNF-α and LTB4 (Canetti and others 2003). Additionally, mature IL-18 is produced by IL-15-stimulated RA synovial fluid neutrophils but not by neutrophils from healthy individuals (Verri and others 2007).
The migration of monocytes has also been shown to be mediated both directly and indirectly by IL-18. Initially, IL-18 stimulation of RA synovial fibroblasts was shown to induce the production of monocyte chemoattractant, MIP-3α/CCL20) (Ruth and others 2003). Subsequently, IL-18 at concentrations found in RA synovial fluid was shown to directly induce both monocyte migration in vitro and in vivo using an RA synovial tissue severe combined immunodeficiency (SCID) mouse chimera (Ruth and others 2010). IL-18-induced migration of monocytes was shown to be dependent upon signaling through p38 and ERK1/2 pathways (Fig. 2).
TNF-α is an important cytokine in the pathogenesis of RA. TNF-α is produced by RA synovial macrophages and fibroblasts. TNF-α has the ability to induce the production of IL-18, IL-18BP, and caspase-1 by RA synovial fibroblasts. Regulation of all 3 of these proteins is necessary to control the amount of biologically active free IL-18. Recently, the amount of free IL-18 (biologically active IL-18 not bound by IL-18BP or soluble IL-18R) was determined to be greater in RA synovial fluid than in osteoarthritis (OA) synovial fluid (Shao and others 2009; Marotte and others 2010). This is due to the paucity of IL-18BP in RA synovial fluid relative to the amount in OA synovial fluid. Interestingly, activation of the ERK1/2 signaling pathway is required for the production of IL-18 by RA synovial fibroblasts; however, it is not involved in the production of IL-18BP. TNF-α stimulation of RA synovial fibroblasts induces IL-18BPa via activation of c-Jun N terminal kinase (JNK) and NFkB, which act upstream of IRF-1 activation (Morotte, unpublished). As a result inhibition of the ERK1/2 using a specific inhibitor reduces the level of bioactive IL-18. In vitro treatment of RA synovial fibroblasts with IL-18 does not affect the production of IL-18BP. However, experiments with exogenous IL-18BP-Fc caused a reduction of ERK1/2 signaling in TNF-α-stimulated RA synovial fibroblasts in vitro. This finding suggested that IL-18BPa may be an important regulator of TNF-α-induced IL-18 expression by RA synovial fibroblasts and as such is a potential target for future therapies (Marotte and others 2010).
Angiogenic Properties of IL-18
We initially identified IL-18 as an angiogenic mediator that can directly induce endothelial cell migration in vitro and blood vessel formation in vitro and in vivo (Park and others 2001; Amin and others 2010). These effects were independent of other angiogenic mediators, including VEGF, SDF-1α/CXCL12 or TNF-α, suggesting that IL-18 has direct angiogenic properties. Accordingly, IL-18 gene-deficient mice have reduced angiogenesis as determined by the in vivo Matrigel plug assay (Amin and others 2010). Complicating matters, others have found IL-18 to have antiangiogenic properties. Specifically, IL-18 was shown to inhibit fibroblast growth factor-2 (FGF-2)-induced endothelial cell proliferation in vitro and FGF-induced corneal revascularization in vivo (Cao and others 1999). IL-18 was also shown to induce cardiac endothelial cell death (Chandrasekar and others 2006).
RA is a chronic inflammatory disease in which the proliferation and development of blood vessels is critical for both the development and maintenance of the synovial pannus. The angiogenic properties of RA synovial tissue were shown to be in part due to IL-18, as RA synovial tissues inserted into SCID mouse induced greater HMVEC migration in response to IL-18 than normal synovial tissue in this model. Recently, we showed that IL-18-stimulated HMVEC migration and in vitro tube formation involves Src and JNK phosphorylation (Amin and others 2010).
Additionally, IL-18 induces RA synovial fibroblasts to produce angiogenic mediators, including SDF-1α/CXCL12, MCP-1/CCL2, and VEGF (Cho and others 2006; Amin and others 2007). RA synovial fibroblasts stimulated with IL-18 signal through p38, NFκB via JNK2 and protein kinase C δ (PKCδ), activating transcription factor-2 via JNK2, and Akt via PI3K to induce SDF-1α (Fig. 2). Similarly, IL-18 stimulation results in MCP-1/CCL2 expression through PI3K activation of Akt and JNK2 activation of NFκB; however, PKCα, not PKCδ, is involved in NFκB activation. Finally, VEGF expression in RA synovial fibroblasts was dependent upon IL-18 stimulation, resulting in either nuclear translocation of NFκB due to JNK2 and PKCα or activation of the AP-1 signaling pathway (Cho and others 2006; Amin and others 2007).
IL-18 as a Target for Future RA Therapy
The reduction of IL-18 has been an objective of several treatments for immunological diseases, including RA. Given the complex regulation of biologically active IL-18, there are several potential targets that can result in its reduction. Potential tools in reducing IL-18 levels have included IL-18 neutralizing monoclonal antibodies, P2X7 antagonists, caspase-1 inhibitors, soluble IL-18R, IL-18R blocking monoclonal antibodies, IL-18BP, and inhibitors targeting IL-18R signaling pathways (Fig. 3). In the mouse CIA model, administration of either anti-IL-18 or recombinant IL-18BP reduced the clinical severity of progressing arthritis through altering the cytokine balance (Plater-Zyberk and others 2001). Later, a gene therapy approach with adenoviral IL-18BPc reduced inflammation and joint destruction in mouse CIA (Smeets and others 2003). Taking this target one step further, an adenovirus containing IL-18BP and IL-4 fusion gene was able to alter the immune response in mouse CIA through increasing Th2 cytokines while, at the same time, reducing the level of Th1 cytokines (Leng and others 2008). Recombinant human IL-18BP was tried clinically for the treatment of active moderate to severe RA and has been reported to be safe at the highest dosage over a 6-week subcutaneous treatment regiment (Tak and others 2006). However, therapeutic efficacy of this approach was not confirmed.
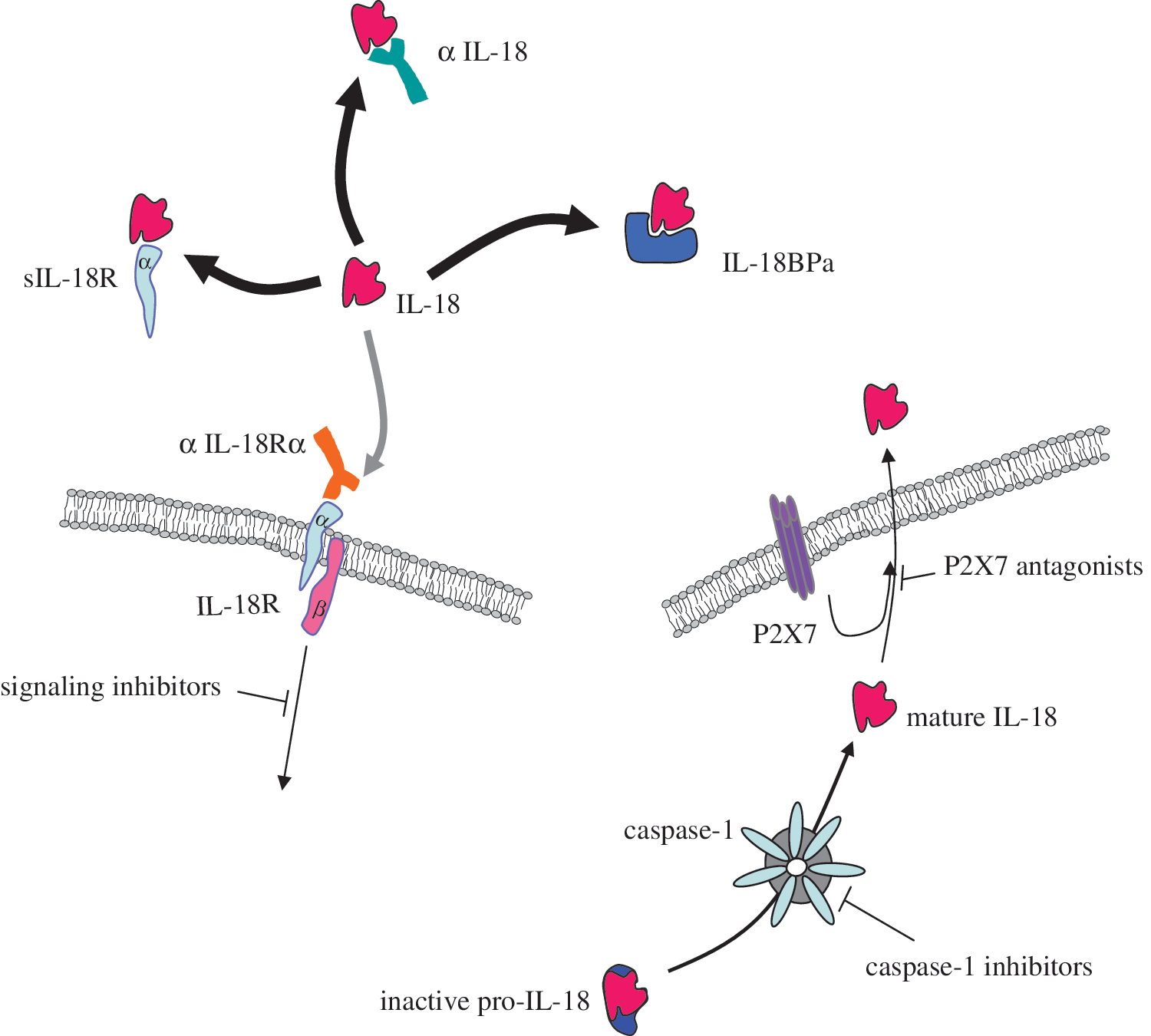
Targets for IL-18-directed therapy in rheumatoid arthritis. α IL-18, anti-IL-18 neutralizing antibody; αIL-18Ra, anti-IL-18 receptor a; IL-18BPa, IL-18 binding protein a; P2X7, purinergic receptor P2X7; sIL-18R, soluble IL-18 receptor.
Adenovirus encoding a natural soluble IL-18R β chain reduced IL-18-induced IFN-γ, IL-4, and IL-17 production by mouse T cells in vitro, suggesting that it functionally inhibits IL-18 stimulation; however, when administered in vivo in the mouse CIA model, it exacerbated arthritis and increased the amount of cartilage and bone erosion (Veenbergen and others 2010). More promising are caspase-1 inhibitors that in addition to preventing the conversion of pro-IL-18 to active mature IL-18 have the added effect of also inhibiting IL-1 activity. A couple of different caspase-1 inhibitors have been developed; pralnacasan was the first one to undergo clinical testing. Phase II clinical trials showed that it significantly ameliorated RA; however, later trials were stopped due to liver toxicity problems (Siegmund and Zeitz 2003). Another specific caspase-1 inhibitor, VX-43198, inhibited the release of IL-18 from human peripheral blood mononuclear cells in vitro and its parent molecule V-765 reduced arthritis when given orally to CIA mice (Wannamaker and others 2007). Any of these proposed therapies may prove to be a useful treatment; however, they will need to be specific so as to not excessively immunosuppress the patient.
Summary
IL-18 is a very potent mediator of inflammation in RA. Its activity is highly regulated by controlling its expression, translation, enzymatic cleavage, and release. In addition, there are both soluble IL-18R and IL-18BP that can bind to active IL-18 and sequester it preventing its functional activity. Finally, IL-18-initiated cytoplasmic signaling cascades are responsible for initiating changes in gene expression and cell behavior. In the RA joint biologically active IL-18 is produced by synovial macrophages and synovial fibroblasts in a positive feedback loop in which TNF-α from synovial macrophages stimulates synovial fibroblasts to release IL-18, which in turn stimulates synovial macrophages to release more TNF-α. In the RA joint, IL-18 induces leukocyte recruitment through upregulating endothelial adhesion molecules and by directly functioning as a neutrophil, macrophage, and T lymphocyte chemoattractant and indirectly by stimulating synovial fibroblasts to release chemokines. Finally, IL-18 functions as an angiogenic mediator directly increasing synovial blood vessel formation and indirectly by inducing synovial fibroblasts to produce angiogenic chemokines and other angiogenic mediators.
Conclusion
Targeting future therapies at regulating IL-18 biological activity is very promising. The parallels between IL-18 and the IL-1β activation process make targeting caspase-1 inhibitors and P2X7 antagonists very attractive because one can potentially inhibit both inflammatory mediators simultaneously. The naturally occurring IL-18BP and soluble IL-18R are also prime targets because of their low potential toxicity. Neutralizing anti-IL-18 or anti-IL-18R monoclonal antibodies are also good potential future therapies as they would have high affinity for their target antigens. Finally, targeting the signaling pathways that IL-18 activates is a potentially novel therapeutic strategy for the treatment of RA.
Footnotes
Acknowledgments
This work was supported by the Frederick G.L. Huetwell and William D. Robinson, M.D. endowment and funds from the Veteran's Administration Research Service.
Author Disclosure Statement
No competing financial interests exist.