
Abstract
Rheumatoid arthritis (RA), an autoimmune disease causing inflammation, destruction, and deformity of the joints, affects around 1% of the world population. It is a systemic disease as patients exhibit extra-articular manifestations as well. Collagen-induced arthritis (CIA) in DBA/1 mice is one of the many animal models used to study possible pathogenic mechanisms of RA. It involves immunizing mice with collagen type II in complete Freund's adjuvant. Here we briefly review the general characteristics of RA and CIA and present an overview of data obtained by studying CIA in several gene knockout mice. In particular, detailed analysis of CIA in interferon-gamma (IFN-γ) receptor-deficient mice has pin-pointed IFN-γ as an important cytokine in the pathogenesis and has exposed new functions of IFN-γ in immunological processes. Pilot trials with exogenous IFN-γ in RA have been indicative of a beneficial effect. That improvement of the disease symptoms by IFN-γ treatment was not spectacular may be explained by the fact that RA is a heterogeneous disease in which the severity of the autoimmune disease is strongly determined by environmental factors.
Rheumatoid Arthritis
Genes associated with RA risk
Certain class II major histocompatibility complex (MHC) haplotypes predispose to RA. In all ethnic populations, the presence of a shared epitope in certain HLA-DRB1 alleles augments susceptibility to disease; the epitope may influence auto-antigen presentation by interacting with T cells or with the antigen itself (Gregersen and others 1987). In European study populations, a polymorphism in the PTPN22 gene has been found to be associated with risk for RA development, whereas in Asians the polymorphism does not exist. PTPN22 is a tyrosine phosphatase that exerts negative feedback in T cell receptor signaling. An association found in Japanese but not in Caucasian populations is with a polymorphism in the PADI4 gene that codes for peptidylarginine deiminase 4, an enzyme responsible for citrullination of proteins. Antibodies directed against citrullinated antigens are a hallmark of RA, suggesting that the enzyme plays a role in immunopathogenesis. Other recently identified candidate genes for influencing RA risk are those coding for STAT4 and CD40 (Goronzy and Weyand 2009).
Studies indicative of an association between RA risk and polymorphisms in the gene encoding interferon-gamma (IFN-γ) (Khani-Hanjani and others 2000; Vandenbroeck and others 2003) have not been confirmed in a subsequent study (Constantin and others 2001). Likewise, no association could be demonstrated with polymorphisms in the IFN-γ receptor 1 (IFN-γR1) gene (Mattyasovszky and others 2006).
Despite evidence for substantial genetic predisposition for RA, the concordance rate in monozygotic twins is only 15%. Therefore, it is generally assumed that genetic factors account for only 50% of the risk to develop RA, and that disease incurrence depends also on posttranslational events elicited by environmental factors.
Environmental risk factors
Bacterial and viral infections have been linked with initiation of polyarthritis in genetically susceptible subjects (Hyrich and Inman 2001). Some of the proposed mechanisms to explain the link are molecular mimicry, epitope spreading, and the expression of superantigens by the pathogen. Likewise, intestinal commensal bacteria were linked with the development of autoimmunity (Lee and Mazmanian 2010). According to a recent report, a specific bacterium, Porphyromonas gingivalis, which is a major player in peridontitis, may be a causal factor of RA. It is proposed that citrullinated α-enolase plays a central role. P. gingivalis infection causes citrullination of proteins in the gingiva, which in combination with danger signals results in the activation of a citrulline-specific pathogenic T cell response, followed by B cell activation and production of antibodies to citrullinated protein antigens (ACPA). In the joint, a second inflammatory event leads to citrullination of joint proteins. Circulating ACPA can form immune complexes in the joint, which results in perpetuation of the local inflammation leading to chronic RA (Lundberg and others 2010).
Another well-recognized risk factor for RA is smoking. Klareskog and others (2008) recently hypothesized that smoking may cause citrullination of proteins in the lungs and that, at the same time, components in the smoke act as immunoadjuvants. The subsequent antibody response to ACPA would cause synovitis. Likewise, exposure to traffic-mediated air pollution has been reported to augment the risk for developing RA (Hart and others 2009). However, the underlying mechanism remains to be elucidated.
Autoimmune nature of RA
Approximately 70% of RA patients have rheumatoid factor (RF) in their serum, that is, antibodies directed against the Fc portion of immunoglobulines of the IgG class. RF can form immune complexes that are deposited in arthritic synovium and activate complement, thereby reinforcing B cell activation and perpetuating an Fc-receptor-mediated positive-feedback loop (Edwards and Cambridge 2006). A positive RF reaction is considered a characteristic, although not specific, hallmark of the disease. Recently, another kind of autoantibody, namely, ACPA, has been identified as a more specific serologic marker of RA. In fact, the previously mentioned HLA-DRB1 shared epitope risk alleles are associated only with a subset of RA that is ACPA or RF positive, indicating that multiple disease subsets exist (Klareskog and others 2008). In addition to these diagnostic antibodies, autoantibodies directed to cartilage components, such as collagen type II (CII), have been described in RA patients (Nandakumar 2010).
Besides a humoral response, the pathologic process in RA is composed of a cellular component. The initial trigger involves both innate and adaptive immunity. The innate immune response consisting of macrophages, dendritic cells, and synovial fibroblasts initiates a cascade of cytokine and chemokine production to attract T and B cells to the site of injury. Through antigen presentation, an antigen-specific immune response is generated. Moreover, infiltrating neutrophils can produce enzymes that damage the synovium. Together with the immune complexes formed by antibodies, this will ultimately lead to destruction of the joint (McInnes and Schett 2007). Since joint pathology in RA is very similar to that in collagen-induced arthritis (CIA), an animal model for RA, it will be discussed in more detail in following sections.
CIA in Mice as a Model for RA
Among several animal models for RA, CIA in DBA/1 mice is the most used. In 1977, Trentham and others first reported that immunization of rats with an emulsion of human, chick, or rat CII in complete Freund's adjuvant (CFA) resulted in the development of an erosive polyarthritis associated with an autoimmune response against cartilage (Trentham and others 1977). Later, other research groups described similar protocols for induction of CIA in mice (Courtenay and others 1980) and nonhuman primates (Cathcart and others 1986). Since CII is the major constituent protein in articular cartilage, the immune response that develops after immunization with CII will target the joints. One of the aspects of this immune response is the production of CII-specific antibodies, a feature that has also been reported in RA (Nandakumar 2010). As in human RA, mice immunized with CII also produce RF (Tarkowski and others 1989). Another pathologic feature that characterizes both RA and CIA is the pronounced synovitis with accompanying cartilage degradation and bone erosions. As is the case with RA, susceptibility to CIA in mice is associated with the expression of specific class II MHC molecules, which in mice is referred to as the H-2 complex. In mice, strains expressing H-2q (eg, DBA/1 and B10.Q mice) or H-2r (eg, B10.RIII mice) are susceptible for CIA (Wooley and others 1981, 1985).
Although being largely similar, RA and CIA also differ in some respects. For instance, joint pathology in RA is chronic and symmetric, whereas CIA in mice is nonsymmetric and transient. Whereas RA is generally considered by clinicians as a systemic disease with prominent articular symptoms, experimentalists working with CIA have so far largely neglected possible systemic aspects of the disease. However, as evident from our recent data, extra-articular manifestations, that is, pulmonary pathology, do occur in CIA especially in the absence of IFN-γ (Schurgers and others, submitted). The similarities and differences between human RA and CIA are summarized in Table 1.
Nonexhaustive list of similarities and differences in the clinical disease course and pathologic mechanisms between RA and CIA.
We recently demonstrated pulmonary manifestations in IFN-γR KO mice with CIA, challenging this statement (Schurgers and others, submitted).
CII, collagen type II; CIA, collagen-induced arthritis; IFN-γR; interferon-gamma receptor; MHC, major histocompatibility complex; RA, rheumatoid arthritis.
Comparison between an unaffected and an arthritic joint
Joints are divided into 3 structural groups: fibrous, cartilaginous, and synovial joints. RA and CIA target the synovial joint (or diarthrosis). The ends of the 2 bones that make contact in a synovial joint are covered by hyaline cartilage. The joint is tightly held together by a fibrous capsule, lined at its inner side by the synovial membrane. The synovial cavity is filled with synovial fluid, produced by the cells of the synovial membrane. The highly viscous fluid lubricates the joint and provides nutrition for the articular cartilage (Hamerman and others 1970). Articular cartilage is composed of a complex mixture of water, proteoglycans, and 5 types of collagen (CII, CVI, CIX, CX, and CXI). CII is the most abundant type, making up 80%–85% of the total. Chondrocytes, the only cells present in the cartilage, are responsible for the maintenance of the tissue by producing the proteins of which cartilage consists (Cohen and others 1998; Cremer and others 1998).
Two structures make up the synovium, the intimal lining layer, which is composed of 1–3 cell layers, and the subintima, which merges with the joint capsule. The intimal lining layer consists of 2 cell types: the macrophage-like synoviocytes (or type A cells) and the less abundant fibroblast-like synoviocytes (FLS) (or type B cells). Under normal conditions, the subintima is relatively hypocellular and consists of fibroblasts intermixed with small blood vessels, lymphatic vessels, and fat cells (Hamerman and others 1970; Scott and others 2010).
Joints affected by RA or CIA suffer devastating consequences for their normal organization. The normally hypocellular synovium becomes infiltrated with immune cells (T cells, B cells, macrophages, and neutrophils). This leads to formation of a pannus, a hyperplastic membrane of synoviocytes that exhibits a tissue-invasive character, targeting bone and cartilage. Production of matrix-degrading enzymes results in the loss of cartilage, whereas the formation of osteoclasts induces bone erosion (Goronzy and Weyand 2009). The synovial fluid, which is normally acellular, becomes highly infiltrated, predominantly with neutrophils. All these changes result in reduced functioning of the joint accompanied by pain and stiffness.
Role of cellular components and associated cytokines in RA and CIA
T cells
In the inflamed synovial membrane of RA patients, T cells make up 75%–90% of the lymphocytic infiltrate (Bankhurst and others 1976). This, together with the association of RA with class II MHC genes, allows for speculation that T cells play a major role in the pathogenesis. Experimental evidence comes from observations that administration of CII-specific T cells can induce arthritis in naïve mice (Holmdahl and others 1985). Moreover, mice deficient for CD4 are less susceptible to CIA than wild-type mice. Deficiency for CD8 did not affect susceptibility to CIA (Ehinger and others 2001). When, in the 1990s, the Th1/Th2 paradigm became the basis for classification of immune responses, both RA and CIA were initially considered to be Th1-driven diseases. The main reason for this view was that the cellular infiltrate in the inflamed synovia was reminiscent of that seen in delayed-type hypersensitivity (DTH)-mediated inflammation. However, studies on the role of endogenous IFN-γ, the prototype Th1 cytokine, yielded conflicting results. Mice lacking a functional IFN-γR were found not to be protected but rather to develop a more severe form of CIA (Manoury-Schwartz and others 1997; Vermeire and others 1997). More recently, Th17 cells, producing cytokines such as interleukin (IL)-17 and IL-22, were implicated as major players in the pathogenesis of CIA (Murphy and others 2003). The conflict was resolved after the discovery of an additional functional category of helper T cells, the Th17 cells. Th17 cells differentiate from naïve T cells through stimulation by transforming growth factor β (TGF-β), IL-6, and IL-23, but their differentiation is suppressed by IFN-γ (Zhu and others 2010). IL-17 promotes inflammation by enhancing the production of proinflammatory cytokines tumor necrosis factor alpha (TNF-α), IL-1β, IL-6, and receptor activator of NF-κB ligand (RANKL) (Peck and Mellins 2009). Supporting the hypothesis that Th17 cells are more important than Th1 cells in the pathogenesis of RA is the fact that IL-17 can be readily detected in RA synovium (Chabaud and others 1999), whereas IFN-γ is present at low levels in RA synovium and synovial fluid (Firestein and Zvaifler 1987; Simon and others 1994). Further evidence for a role of Th17 cells in CIA is provided by studies in which neutralization of endogenous IL-17 by antibodies (Lubberts and others 2004; Kelchtermans and others 2009) or by knocking out the IL-22 gene (Geboes and others 2009) protects against the disease.
B cells
Besides production of autoantibodies such as RF, ACPA, and anti-CII antibodies, B cells can contribute to the pathogenesis of RA and CIA by producing cytokines, such as IL-6, or by serving as antigen-presenting cells (APCs) to activate antigen-specific T cells. A role for B cells in antigen presentation is supported by the observation that B cell depletion prevents the formation of ectopic germinal centers in the synovium, resulting in suboptimal activation of T cells (Takemura and others 2001). It was even demonstrated that CIA could not be induced in mice deficient for B cells (Svensson and others 1998). Indeed, targeting B cells has proven to be a very successful therapeutic option in the treatment of RA. Rituximab, a CD20-specific monoclonal antibody, depletes all B cell subsets (except plasma cells), which results in a significant clinical benefit in RA patients (Edwards and others 2004).
Neutrophils
Neutrophils normally function as a first line of defense against invading pathogens. In a joint affected by RA, large numbers of neutrophils can be found in the synovial fluid, where they are activated by immune complexes, cytokines, or complement. Besides secretion of a wide variety of cytokines, including IL-1β, IL-6, and TNF-α, neutrophils produce high levels of reactive oxygen species and matrix-degrading enzymes such as matrix metalloproteinase 9 (MMP9 or gelatinase B), causing cartilage destruction. Neutrophils also express PADI4, which, after apoptosis of these short-lived cells, can cause citrullination of cartilage proteins, thereby generating epitopes recognized by ACPA and perpetuating the disease (Edwards and Hallett 1997; Cascao and others 2010). In several animal models, depletion of neutrophils prevented the development of arthritis (Wipke and Allen 2001; Tanaka and others 2006), as did the blockage of granulocyte chemotactic protein-2 (GCP-2), the most important neutrophil chemokine in the mouse (Kelchtermans and others 2007). These data clearly indicate a role for neutrophils in the pathogenesis of RA and CIA.
Macrophages and osteoclasts
In joints affected by RA, the number of synovial macrophages correlates with articular destruction (Mulherin and others 1996). Synovial macrophages and attracted monocytes are involved in cytokine and chemokine production, matrix degradation, and angiogenesis. Activation of these macrophages likely occurs through immune complexes/FcγRs and ligation of toll-like receptors (TLRs), recognizing various microbial products as well as putative endogenous ligands. Evidence for involvement of TLRs in the pathogenensis of RA comes from demonstration that inhibition of TLR2 and TLR4 in rodent models of RA reduces the development of arthritis (Joosten and others 2003; Abdollahi-Roodsaz and others 2007). Among the abundance of cytokines produced by activated macrophages, TNF-α, IL-1β, and IL-6 are the most important.
TNF-α can be detected in synovium and synovial fluid (Chu and others 1991) and the importance of this cytokine is reflected by the spontaneous development of arthritis in TNF-α transgenic mice that overexpress a modified human TNF-α transgene (Keffer and others 1991). Among other functions, TNF-α induces production of other proinflammatory cytokines, activates polymorphonuclear cells, and enhances cartilage breakdown (Zwerina and others 2005).
IL-1β can be readily detected in serum of RA patients and its levels correlate well with disease severity. Mice deficient for the IL-1 receptor antagonist, a naturally occurring counter regulatory cytokine, develop a spontaneous inflammatory arthritis (Horai and others 2000), indicating the importance of this cytokine. IL-1β exerts its proinflammatory effects through stimulation of cytokine and chemokine production and matrix-degrading enzymes.
High levels of IL-6 can be detected in synovial fluid of RA patients (Houssiau and others 1988) where it can exert its functions such as terminal differentiation of macrophages, acting as a growth factor for T cells and enhancing B cell proliferation and subsequent antibody production (Zwerina and others 2005). Blocking the IL-6 receptor in mice with CIA resulted in inhibition of arthritis development (Takagi and others 1998). The most convincing evidence that TNF-α, IL-1β, and IL-6 are key regulators of RA comes from new therapies targeting these cytokines. They have all been proven to be successful and are now being used in the clinic.
Under the influence of RANKL, produced by activated T cells and synovial fibroblasts, cells of the monocyte/macrophage lineage can differentiate and fuse into multinucleated osteoclasts. These osteoclasts erode bone adjacent to the joint, causing irreversible damage (Schett 2007). TNF-α can induce osteoclastogenesis in a RANKL-independent manner (Abu-Amer and others 2000).
Synovial fibroblasts
While osteoclasts are the sole bone-eroding cells, FLS are the main degraders of cartilage. The pannus is mainly composed of FLS, which, in RA, possess invasive potential not present in other fibroblasts. After stimulation with cytokines such as IL-1β, IL-6, and TNF-α, FLS secrete MMPs (for instance MMP1, a collagenase), aggrecanases, and cathepsins to degrade cartilage, and secrete more cytokines and chemokines. For instance, FLS produce chemokines to attract T cells [macrophage inflammatory protein-1α (MIP-1α), SDF-1, regulated on activation, normal T cell-expressed and secreted (RANTES)], neutrophils (IL-8), and monocyte/macrophages [inducible protein-10 (IP-10), monocyte chemoattractant protein-1 (MCP-1)], thereby creating a mix of cells capable of initiating/perpetuating the synovial inflammation. The effector functions of FLS are extensively reviewed in reference (Bartok and Firestein 2010).
The Role of IFN-γ in Synovial Inflammation
IFN-γ is produced mainly by NK cells and a particular subset of T cells, namely, Th1 cells. Although at first it was believed that these cell types were the sole producers of IFN-γ, more recent data indicated that professional APCs, B cells, and NKT cells can also produce IFN-γ (Frucht and others 2001). APCs also produce 2 potent inducers of IFN-γ, namely, IL-12 and IL-18. A functional IFN-γR consists of 2 ligand-binding IFNGR1 chains and 2 signal-transducing IFNGR2 chains. The receptor signals through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway to induce IFN-γ-regulated gene transcription (Schroder and others 2004).
Cellular effects of IFN-γ
Macrophage activation
Soon after its discovery, IFN-γ has been recognized as one of the most potent activators of macrophages (Billiau 1996; Billiau and Matthys 2009). Besides stimulating antigen-presentation, IFN-γ induces a heightened microbicidal state in macrophages: IFN-γ-primed macrophages react more readily to danger signals such as lipopolysacharide or bacterial DNA, which are sensed through TLRs. Also, expression of genes involved in the formation of reactive oxygen or reactive nitrogen species is augmented, thereby aiding in elimination of pathogens (Boehm and others 1997; Schroder and others 2004). IFN-γ induces largely the same effects in neutrophils (Ellis and others 2004).
Antigen presentation
IFN-γ upregulates expression of MHC-II molecules on APCs. Thus, it enhances the potential of cells to present exogenous antigens to CD4+ Th cells or CD8+ cytotoxic T cells in both quantitative and qualitative ways (Schroder and others 2004). Further, IFN-γ can also upregulate expression on APC of co-stimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) (Boehm and others 1997). However, IFN-γ has also been shown to induce MHC-II expression in cells normally not expressing this molecule. Therefore, it might support development of autoimmunity by stimulating the presentation of auto-antigens to autoreactive T cells, resulting in breakdown of peripheral tolerance (Billiau and Matthys 2009).
Development of T helper cells
Naïve T cells whose receptor recognizes antigenic peptide/MHC-II complexes on APCs differentiate into one of several T helper categories, depending on the prevalent cytokine environment. In the presence of IFN-γ, together with IL-12 produced by APCs, differentiation takes the Th1 direction. In turn, these Th1 cells produce IFN-γ, which acts as a positive feedback loop to further stimulate Th1 differentiation of other naïve T cells. Moreover, IFN-γ inhibits differentiation of naïve T cells into Th2 or Th17 cells. By this mechanism, IFN-γ can counteract production of IL-4, produced by Th2 cells, which itself is a potent inhibitor of Th1 differentiation (Zhu and others 2010).
Chemokine induction and leukocyte trafficking
Correct trafficking of leukocytes to a site of inflammation is crucial for immune responses to be effective. This trafficking is largely governed by chemokines, a variety of which can be induced by IFN-γ, albeit in synergy with TNF-α and IL-1β, for example, IFN-γ-inducible protein 10 (IP-10), MCP-1, MIP-1α/β, monokine induced by gamma interferon (MIG), or RANTES (Gouwy and others 2005; Loos and others 2006; Proost and others 2006). These chemokines are chemotactic for monocytes and lymphocytes. Extravasation of leukocytes is mediated through interaction of adhesion molecules expressed on endothelial cells and leukocytes. IFN-γ strongly induces the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells, thereby enhancing leukocyte recruitment to the site of inflammation (Schroder and others 2004).
Role of IFN-γ in the pathogenesis of CIA
Endogenous IFN-γ acts as a disease-limiting factor in CIA
The protective role of IFN-γ in classical CIA in mice was established by administration of IFN-γ or neutralizing anti-IFN-γ antibody, or by using genetically engineered mice with defects in production or action of IFN-γ (Nakajima and others 1990; Williams and others 1993; Manoury-Schwartz and others 1997; Vermeire and others 1997; Guedez and others 2001). Kelchtermans and others (2008) have extensively reviewed these studies as well as studies identifying the different underlying mechanisms. Figure 1 provides an overview of the cellular effects of IFN-γ in experimentally induced arthritis.
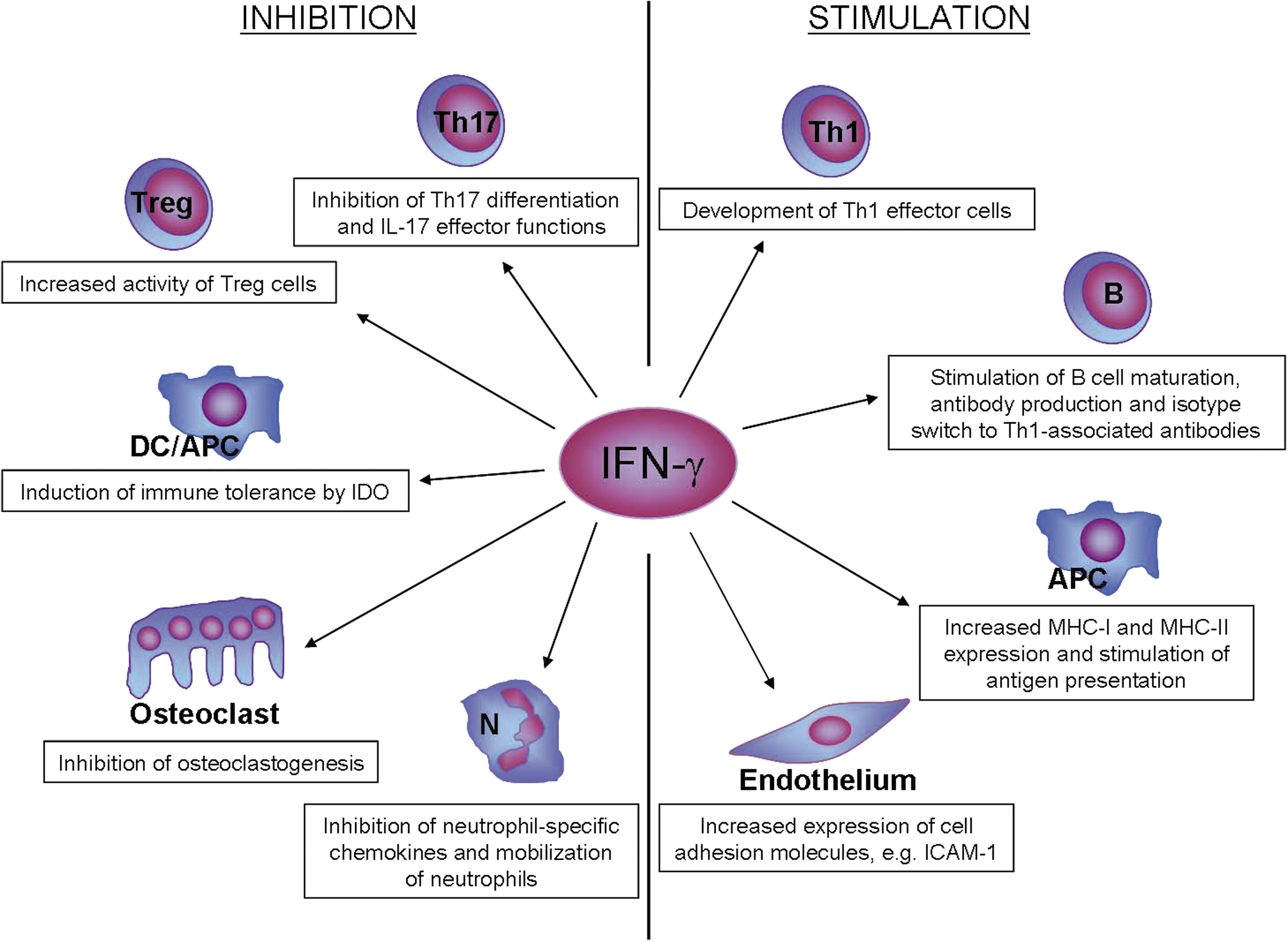
Disease-limiting (left) and disease-promoting (right) activities of IFN-γ in the pathogenesis of experimentally induced arthritis. This figure summarizes how a single cytokine, IFN-γ, can function both as an inducer and a regulator of immune responses. For details, see text. APC, antigen-presenting cell [including macrophages and dendritic cells (DC)]; IFN-γ, interferon-gamma; N, neutrophil; IDO, indoleamine 2,3-dioxigenase; ICAM-1, intercellular adhesion molecule-1; MHC, major histocompatibility complex.
IFN-γ inhibits differentiation of monocyte/macrophages into osteoclasts through ubiquitin-proteasome-mediated degradation of TNF receptor-associated factor-6 (TRAF6), a signal molecule crucial for osteoclast development (Takahashi and others 1986). Moreover, as apparent from studies with induction of CIA in IFN-γR knockout (KO) mice, IFN-γ interferes with the accompanying boost in splenic myelopoiesis and thereby reduces the number of available osteoclast precursors (De Klerck and others 2004). Further, in an antigen-induced arthritis model, IFN-γ has also been found to inhibit IL-1β-mediated production of MMP1 and MMP3 by synovial fibroblasts, thereby limiting cartilage degradation (Page and others 2010).
An additional protective mechanism of IFN-γ is by stimulating regulatory T (Treg) cells, which play a major role as downregulators of CIA (Kelchtermans and others 2005; Morgan and others 2005). IFN-γ facilitates development of Treg cells from precursor T cells (Wang and others 2006) and also makes them more suppressive during CIA (Kelchtermans and others 2005). Conversely, IFN-γ inhibits the development of Th17 cells and suppresses their effector functions (Kelchtermans and others 2009; Zhu and others 2010). Specifically, in fibroblast cultures, IFN-γ inhibited the IL-17-induced expression of IL-6, RANKL, and GCP-2 (Kelchtermans and others 2009), suggesting that, in vivo, it may reduce inflammation, osteoclast differentiation and neutrophil influx.
Interplay between adjuvant and IFN-γ in CIA
Immunization of IFN-γR KO DBA/1 mice with CII in CFA results in CIA that is characterized by an earlier onset and a more severe disease course as compared to wild-type mice (Vermeire and others 1997; De Klerck and others 2004). Moreover, immunization of IFN-γR KO mice with CFA, thus omitting the antigen CII, gives rise to arthritis in these mice. By contrast, wild-type mice are not susceptible for this adjuvant-induced arthritis (Geboes and others 2007). When, however, the CFA in the immunization schedule (containing CII) was replaced by incomplete Freund's adjuvant, which lacks heat-killed mycobacteria, IFN-γR KO mice were completely protected while wild-type mice developed CIA with an incidence of 25% (Matthys and others 1999). This reduction in clinical symptoms in IFN-γR KO mice coincided with a reduction in cellular immune responses as measured by DTH reaction toward CII and a reduced splenic myelopoiesis as measured by the number of CD11b+ cells (Matthys and others 1999). Moreover, IFN-γ has been demonstrated to be an essential component of the immune response toward viable mycobacteria (Flynn and others 1993; Kamijo and others 1993). These observations indicate that, under the influence of mycobacteria from CFA, a pathogenic pathway is exposed, which is counteracted by IFN-γ. In several other models of autoimmunity that rely on the use of CFA as an adjuvant, a similar disease-limiting role for IFN-γ has been demonstrated. Examples include experimental autoimmune encephalomyelitis (Billiau and others 1988) and experimental autoimmune uveitis (Caspi and others 1994). One could thus speculate that, in the absence of mycobacteria, myelopoiesis is less important for development of experimental disease models and IFN-γ can execute its natural disease promoting properties by activation of macrophages and autoantigen-specific adaptive immune responses. In the presence of mycobacteria, however, myelopoiesis becomes the more important factor for disease development such that its inhibition by IFN-γ results in limitation of the disease (Matthys and others 2000).
However, some remarks can be postulated in respect to the hypothesis stated above. Immunization of DBA/1 mice with glucose-6-phosphate isomerase (G6PI) in CFA following the same procedure as for the induction of classical CIA results in protection of arthritis in IFN-γR KO mice as compared to wild-type mice, indicating a disease-enhancing role of IFN-γ in this model of autoimmune arthritis (Frey and others 2011). This discrepancy was explained by a difference in disease onset in the 2 model systems (G6PI-induced arthritis in wild-type mice starts from day 7 post–immunization, whereas symptoms of CIA start from day 21 onward) combined with a bi-phasic effect of IFN-γ in the disease course of arthritis. See Table 2 for a comparison between G6PI-induced arthritis and CIA or adjuvant-induced arthritis.
The table lists the disease outcome in DBA/1 wild-type and IFN-γR KO mice, challenged with CIA (CII in CFA), Freund's adjuvant-induced arthritis (CFA), and glucose-6-phosphate isomerase (G6PI)-induced arthritis (G6PI in CFA). All mice were bred in the same animal facility and experiments were performed under the same conditions and used the same doses and batches of CFA. The data are a summary of experiments described by Vermeire and others (1997), Kelchtermans and others (2007), De Klerck and others (2004), Geboes and others (2007), and Frey and others (2011).
+, low; ++, moderate; +++, high; NA, not applicable; ND, not determined; CFA, complete Freund's adjuvant; DTH, delayed-type hypersensitivity; KO, knockout.
Treatment options in RA concerning IFN-γ
Due to its immunomodulatory properties, IFN-γ has been considered as a therapeutic for the treatment of RA and clinical trials exploring this possibility have been conducted. Between 1986 and 1998, several studies were performed administering recombinant human IFN-γ to RA patients. These studies varied from open-label to double-blind placebo-controlled trials, but all reported amelioration, whether or not significant, of arthritic symptoms (Seitz and others 1986; Lemmel and others 1987, 1988; Veys and others 1988; Cannon and others 1989, 1993; German Lymphokine Study Group 1992; Machold and others 1992). Beneficial effects of IFN-γ have also been reported after use in juvenile chronic arthritis (Coto and others 1998). The administration of IFN-γ to RA patients has been reported to upregulate the expression of FcγR 1 on circulating neutrophils (Goulding and others 1992) and to reduce the number of circulating B cells without affecting antibody titres (Pincus and others 1990). By contrast, one clinical trial reported amelioration of articular involvement after treatment of RA patients with anti-IFN-γ antibodies (Sigidin and others 2001).
Since the results of the conducted trials treating RA patients with recombinant IFN-γ were disappointing in the sense that it did not result in dramatic amelioration of arthritic symptoms as compared to other biologicals such as anti-TNF-α therapy, it was omitted as a therapeutic.
Conclusions
CIA in mice has proven to be a useful animal model for studying the pathogenesis of RA. There is close resemblance in histopathology as well as in the production of inflammatory mediators such as chemokines, cytokines, proteases, and autoantibodies. The use of KO mice has been a valuable tool to provide insights into the pathogenesis of the model disease. In particular, the use of IFN-γR KO mice has contributed to the exposure of several new pathways in the development of autoimmune arthritis. The early finding that CIA takes a more severe course in mice deficient in IFN-γ or its receptor came as a surprise given that IFN-γ at the time was considered to possess pro-inflammatory properties only. However, in recent years it became increasingly clear that IFN-γ, depending on the immune context, can also acts as an important immune modulator, for instance, by controlling the development and activity of Th17 cells, neutrophil chemotaxis, and extramedullar osteoclastogenesis, and by enhancing the activity of Treg cells. One aspect of the role of IFN-γ in CIA is that it keeps in check the accompanying myelopoiesis driven by the mycobacterial component of Freund's adjuvant.
These studies with CIA invite speculation that endogenous IFN-γ plays an important role in the pathogenesis of RA in humans. The trigger for IFN-γ production may come from the autoimmune reaction itself or from environmental stimuli such as intercurrent infections. Indeed, given the low accordance rate of RA in monozygotic twins, one must assume that nonendogenous factors significantly contribute to the pathogenesis of RA. Further identification of these environmental factors (smoking is a current topic) as well as uncovering the mechanisms by which these factors contribute to the pathogenesis of RA will be important.
Besides CIA, there are other animal models of RA (with different induction schedules or spontaneously developing) and, nevertheless, each of these models is in one way or another reminiscent to RA, illustrating the heterogeneity in etiology of RA. Importantly, differences in the induction schedule can result in completely opposite outcomes regarding the role of a cytokine in the disease process (ie, disease-limiting role of IFN-γ in CIA versus pro-inflammatory role in G6PI-induced arthritis). It will therefore be important to unravel the heterogeneity of RA, and this knowledge may help us to explain the failure of treatments (such as TNF-α inhibitors) in some groups of RA patients. Conducting clinical trials in selected groups of RA patients, rather than in randomly selected patients, will most likely give better results and in that case the treatment of RA with IFN-γ should be reconsidered.
Footnotes
Author Disclosure Statement
No competing financial interests exist.