
Abstract
Infectious disease is frequently cited as a precursor of subsequent autoimmune disease in genetically susceptible hosts. However, the precise mechanisms required for the transition from infection to autoimmunity have not been well defined. We have developed a mouse model of autoimmune myocarditis initiated by infection with Coxsackievirus B3 to trace the cytokine pathways involved. We found that greater production of interleukin-1β (IL-1β) and tumor necrosis factor-α during the early innate response to virus infection is necessary and sufficient to induce a later heart-specific autoimmune disease. Severity of the autoimmune myocarditis is determined by the profile of a number of T helper 1 (Th1) and Th2 cytokines. Th2 responses are especially pronounced in the most severe forms of myocarditis where eosinophils are prominent. The Th1 pathway can lead to infiltration of the heart, but may be dampened by concurrent INF-γ production. Th17 cytokines also contribute to disease, but the signature Th17 cytokine, IL-17A, is not required for cardiac inflammation. Rather, IL-17A is needed for progression to dilated cardiomyopathy. These findings may provide useful markers to identify individuals prone to develop an autoimmune sequel after infection and suggest future early interventions.
Introduction
In this essay, we deal with these and related issues in reference to a murine model of myocarditis developed in our laboratory. It summarizes the work of a number of students and postdoctoral fellows over the last 3 decades. Many other groups have contributed importantly to the field and their work was described elsewhere (Afanasyeva and Rose 2004).
The Myocarditis Model
To study in detail the mechanisms by which infection can possibly give rise to autoimmune disease, we developed a model of immune-mediated heart muscle inflammation in mice (Herskowitz and others 1987; Rose and others 1987; Rose and others 1988a). In humans, this disease, myocarditis, is often associated with a prior viral infection. One of the most commonly reported agents is Coxsackievirus B3 (CB3). To produce a model closely replicating the human disease, we infected multiple strains of mice with a cardiotropic strain of CB3 obtained originally from the human case of myocarditis (Wolfgram and others 1985; Rose and others 1987). Since this viral infection is common, but a pathologic autoimmune sequel is uncommon, we reasoned that the pathways leading from infection to autoimmune disease must be under tight genetic control (Rose and others 1988b). We found that all of the strains of mice tested developed an acute myocarditis (Beisel and others 1985; Wolfgram and Rose 1989). Histological evidence of acute inflammation appeared by days 4 or 5 in all of the strains examined. It consisted of small foci of myocardial necrosis associated with a primarily focal mononuclear and polymorphonuclear infiltration. Evidence of increasing cardiac cell injury was evident on days 7 and 9 when the lesions were heavily infiltrated with mixed inflammatory cells. After day 9, the inflammatory process diminished and, in most strains of mice, resolved completely by day 21.
In a few strains of mice where the cardiac inflammation did not recede, its appearance changed significantly. In these strains, there were accumulations of lymphocytes and other mononuclear cells and the beginning signs of fibrosis (Neumann and others 1993). In most strains the disease peaked on day 21. By day 45 there was gradual diminution of focal and interstitial inflammation, but increasing evidence of cardiac cell necrosis and fibrosis. In some strains, this led to a striking picture of dilated cardiomyopathy featuring extensive fibrosis, left ventricular dilatation, and cardiac remodeling.
On the basis of these histopathological changes, we were able to divide the myocarditic process into 3 distinct phases (Rose and others 1986). The first was seen in varying degrees of severity in all of the strains of mice tested and was associated until day 9 with the presence of viable virus in the heart. After day 9, there was, in genetically susceptible mice, a second phase of disease in which no viable virus was recovered. During this stage, the mice developed autoantibodies reactive with heart. More precise analysis showed that a portion of the antibodies reacted specifically with cardiac myosin and failed to react with other forms of myosin such as the isoforms found in skeletal muscle or brain (Neu and others 1987a). We concluded that the first early phase of disease found in all strains of mice was viral myocarditis, whereas the second phase observed in only a few strains represented an immunopathic autoimmune response to the prior virus infection. A third phase of the disease became evident at later times in some of the mice and was characterized by physiologic evidence of heart failure and frequently death (Fairweather and Rose 2004). Histologically, this stage of the disease replicated human dilated cardiomyopathy (Fig. 1).
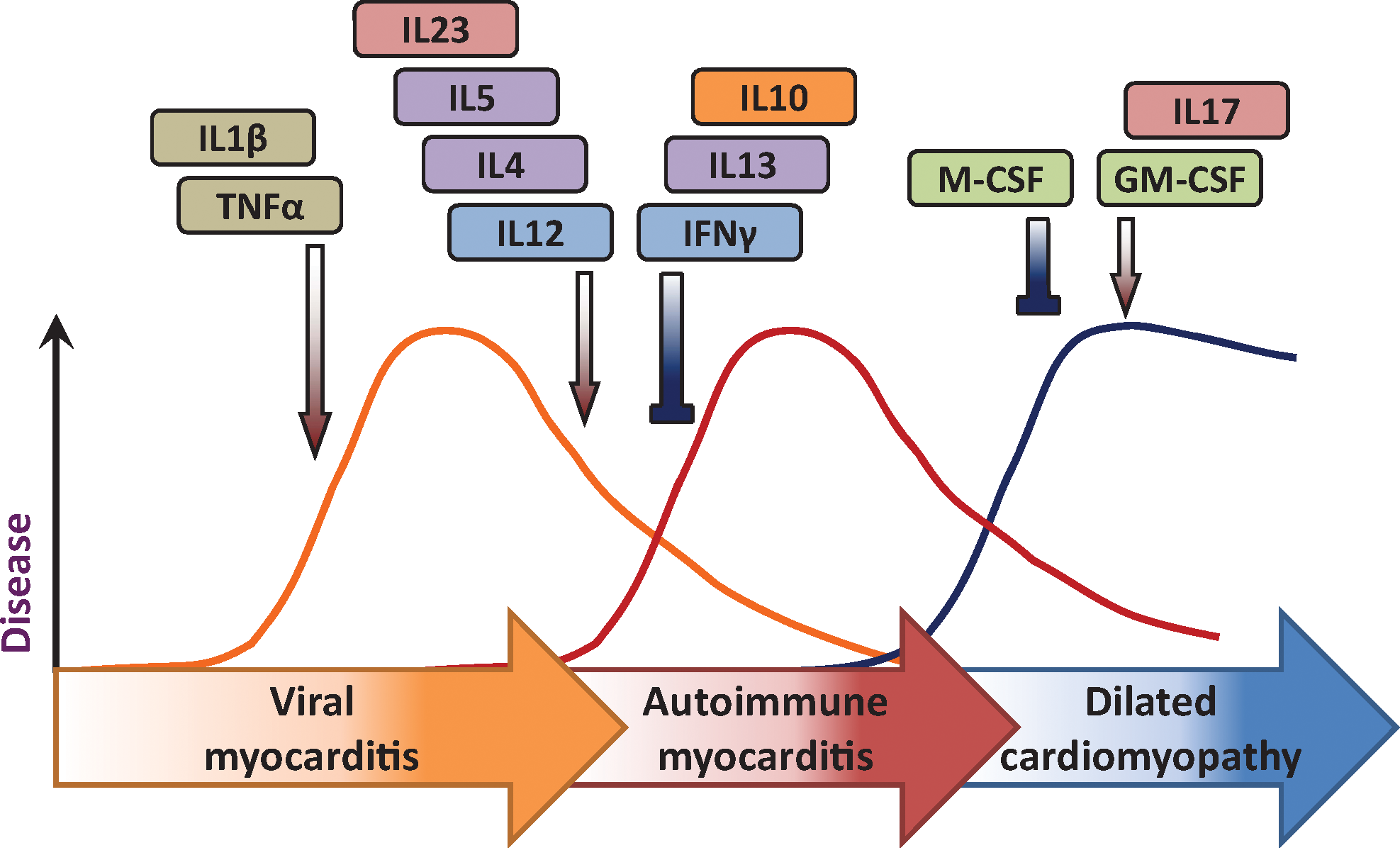
Progression from viral infection to autoimmune heart disease and dilated cardiomyopathy is cytokine regulated.
The model provided us a unique opportunity to study the cytokine pathways that lead from an acute, limited viral infection to immune-mediated inflammatory myocarditis and then to a predominantly fibrotic life-threatening cardiomyopathy. Before proceeding, however, it was necessary to establish firmly that the immune-mediated disease was truly an autoimmune disorder and not dependent on persistent virus. For that purpose, we were fortunate in discovering that the antibodies that characterized the second phase of disease that were specific for cardiac myosin heavy chain (Neu and others 1987a). We, therefore, proceeded to isolate murine cardiac myosin and immunized mice that were susceptible or resistant to the second-phase disease (Neu and others 1987b). We employed skeletal myosin as a control antigen. The results confirmed our suspicion that the second-phase myocarditis was, in fact, autoimmune in origin because only the genetically susceptible animals responded to cardiac myosin with the production of typical lymphocytic monocytic lesions. The strains that were not susceptible to second-phase disease after CB3 infection were unresponsive to cardiac myosin immunization. Skeletal myosin produced no pathologic effects in the hearts of any of the strains examined. Later, we and others were able to complete the circle of evidence by transferring disease from immunized mice to naive recipients with CD4 T cells from immunized donors (Afanasyeva and Rose 2004). Experimental autoimmune myocarditis reproduces an autoimmune disease caused by a virus infection commonly associated with the same disease in humans (Neumann and others 1993; Rose and Hill 1996; Fairweather and Rose 2005).
Infection to Autoimmunity
The model of autoimmune myocarditis described in the previous section allowed us to explore a critical question that had remained unanswered until that time—namely, what are the factors that determine whether an animal (including a human) with an infectious disease will resolve the disease completely or progress to an autoimmune sequela? In our earliest experiments we were able to distinguish some of the genetic characteristics of susceptible and resistant strains of mice (Rose and others 1988b). For example, we found that strains derived from A/J mice were susceptible. A.SW mice were highly susceptible, whereas mice of the B10.S strain derived from C57 BL10 were almost resistant even though they share the same haplotype. By comparing 2 strains of mice, we were able to identify the principal cytokine signals that indicated subsequent susceptibility of mice infected with CB3 to later autoimmune myocarditis (Lane and others 1993). These studies were carried out by James Lane, then a graduate student in the laboratory. He began his experiments by comparing cytokine production during early CB3 infection in susceptible versus resistant mice. We were not surprised to find that there was a major difference in production of 2 of the classical pro-inflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) favoring the susceptible A-strain mice. Equally interesting were follow-up experiments with DeLisa Fairweather, which showed that this difference in cytokine production between resistant and susceptible mice was evident as early as 6 h after CB3 infection (Fairweather and others 2004). These results emphasized the importance of the innate response to the viral infection.
We then proceeded to alter the cytokine profile during the early steps of virus infection. Since lipopolysaccharide (LPS) was known to act as an adjuvant in facilitating production of autoimmune disease, we injected LPS in resistant mice at the time of infection (Lane and others 1991). This combination of virus infection and LPS proved lethal to 70% of the mice by day 15 and resulted in severe diffuse cardiac infiltration with cardiac cell necrosis and increased expression of human leukocyte antigens class II on the myocardiocyte. Antibodies to cardiac myosin were also raised. Injection of LPS alone had no effect.
Other experiments showed that blocking either IL-1β or TNF-α at the time of infection prevented the appearance of autoimmune myocarditis even in susceptible mice (Lane and others 1992). Thus, long-term expression of the IL-1 receptor antagonist effectively marked disease in genetically susceptible A/J mice. Administration of monoclonal antibody to TNF-α or IL-18 also abrogated disease. The finding that inhibiting any one of these cytokines prevented disease suggests that they work in series rather than parallel in producing the transition from infectious to autoimmune myocarditis. To show that the action of these early cytokines was on the autoimmune stage of myocarditis rather than infection, we induced myocarditis by immunization with cardiac myosin. The same effects of the cytokine in blocking development of disease were found.
The final series of experiments was performed by administering recombinant IL-1β and TNF-α to resistant mice (Lane and others 1993). Administration of either IL-1 or TNF separately promoted CB3-induced autoimmune myocarditis in genetically resistant mice. Myocarditis was also observed when uninfected mice were immunized with cardiac myosin in the presence of either IL-1β or TNF-α. These experiments showed for the first time that these early proinflammatory cytokines are both necessary and sufficient to induce the transition from infectious to autoimmune myocarditis and, therefore, are useful early markers of susceptibility.
Cytokine Regulation of the Autoimmune Disease: Downregulatory Cytokines
Experiments were carried out to determine if the autoimmune disease could be prevented even in highly genetically susceptible strain, A/J, by inducing tolerance to cardiac myosin (Wang and others 2000). These experiments were lead by Yan Wang, then a postdoctoral fellow. She found that she could prevent autoimmune myocarditis by nasal installation of cardiac myosin. The procedure reduced the production of a number of important proinflammatory cytokines, including IL-1, IL-2, TNF-α, IL-4, and interferon-γ (IFN-γ). Two downregulatory cytokines, transforming growth factor-β and IL-10 increased, suggesting that the decreased autoimmune disease was due to a heightened suppressor response. Related to induction of regulatory T cells, later experiments by Ziya Kaya and others (2002) showed that IL-10 is a critical cytokine in this form of nasal tolerance, indicating a primary role for induced regulatory T cells. Experiments carried out with Leiping Chen showed that mice deficient in the programmed death ligand (PD-L)1 regulatory factor develop myocarditis spontaneously (unpublished data), illustrating the concept that the development of autoimmune disease may be the consequence of lowered immunoregulation, enhanced immune responsiveness, or both. Shifts in the balance of cytokines can provide clues to the critical cytokine pathways.
Cytokine Regulation of Autoimmune Disease: T Helper 1 Responses
The next series of experiments dealt with the major cytokines classically associated with T helper 1 (Th1) responses in inducing the autoimmune disease. Most of these experiments were carried out using BALB/c mice because they are moderate responders to immunization with cardiac myosin. These animals allow us to measure an increase as well as a decrease in the severity of myocarditic inflammation. These investigations were lead by Marina Afanasyeva, then a graduate student and later a fellow. She found that mice genetically deprived of the β1 chain of major receptor for IL-12 signaling, IL-12R, were fully resistant to the development of myocarditis (Afanasyeva and others 2001b). Activation of signal transducer and activator of transcription (STAT)4 is essential for IL-12 signaling and we demonstrated further that STAT4 knockout (KO) mice were generally resistant to disease, but when it did occur in STAT4 KO mice, the inflammation was rich in eosinophils, a topic that we returned to in later experiments. To confirm directly a role of IL-12 in mediating myocarditis, we treated BALB/c mice with recombinant IL-12 during disease induction and found that it enhanced the cardiac inflammation. These experiments related the induction of lymphocytic myocarditis to Th1 responses.
Because the IL-12 R/STAT4 pathway leads to heightened production of IFN-γ, we next explored the outcome of blocking IFN-γ with an autoantibody (Afanasyeva and others 2001a; Afanasyeva and Rose 2002; Rose and Afanasyeva 2003b). Surprisingly, we found that anti-IFN-γ injection induced exaggerated disease in BALB/c mice, resulting in very severe myocarditis and eventual progression of dilated cardiomyopathy. These experiments showed that IFN-γ and IL-12, 2 classical Th1 cytokines, have opposite effects in myocarditis.
Similar studies were done using CB3-induced disease by Fairweather and others (2005b). This virus-induced form of disease more closely resembles human myocarditis, but the interpretation of results may be complicated by the fact that there is both a viral phase and autoimmune phase of myocarditis. Briefly, we found that signaling through the IL-12 R receptor increased CB3-induced myocarditis as it did in the myosin-induced form of the disease. To investigate the role of IL-12 during the early acute phase of viral myocarditis, BALB/c mice deficient in IL-12 P35 were examined for the development of early phase disease. IL-12 deficiency did not reduce the severity of inflammation at this early time point, indicating that IL-12 is not required for myocardial inflammation itself. Rather, IL-12 deficiency increases the replication of virus in the heart perhaps by causing macrophage and neutrophil depletion. The effect of IL-12 on virus production and inflammation in the heart during CB3 infection operates by reducing IFN-γ.
These experiments clearly showed that IFN-γ, a signature cytokine of the TH1 response, is actually protective in autoimmune myocarditis. Similar findings were reported for 2 other models of autoimmune disease, encephalomyelitis and uveitis. The mechanisms by which IFN-γ regulates autoimmune disease are being investigated by a fellow, Jobert Barin. Using autoimmune myocarditis induced by immunization with the pathogenic peptide of cardiac myosin, he was able to correlate the protective effects of IFN-γ with increased apoptosis of CD4+ T cells over the course of disease. This apoptic effect is embedded in the CD4+ T cell population since it is not dependent upon the stimulating antigen-presenting cell and may represent a form of activation-induced cell death. Yet, even when CD4+ T cells from IFN-γ KO mice were transferred to normal recipients, less disease occurred, suggesting that non-T cell sources of IFN-γ were also available to control disease. It is clear that much more work needs to be done to define the mechanism by which IFN-γ is protective in this and other autoimmune disorders. However, the fact that this cytokine is effective in both reducing viral infection and diminishing the severity of autoimmune disease suggests that agents of this sort may have a useful role in future therapies of human myocarditis where the underlying pathogenic mechanism, virus induced or immune mediated, is frequently unclear (Rose 2009).
Cytokine Regulation of Autoimmune Disease: Th2 Responses
IL-4 is the prototypic TH2 cytokine. These responses are mostly associated with allergic reactions and their role in autoimmune inflammation is often overlooked. A/J mice develop a particularly severe form of experimental myocarditis that is associated with the conspicuous presence of eosinophils and giant cells and a response characterized by high levels of IgG1 antibody to cardiac myosin and upregulation of total IgE (Afanasyeva and others 2001a). Afanasyeva found that blocking IL-4 with anti-IL-4 monoclonal antibody significantly reduced the severity of myocarditis. It was associated with a shift from a Th2-like to Th1-like phenotype as shown by a reduction in the cardiac myosin-specific IgG1 and an increase in IgG2a antibodies. At the same time, there was a dramatic increase in IFN-γ production which, in this case, limits disease. Suppression of IFN-γ is a plausible mechanism by which IL-4 promotes the severity of myocarditis. In brief, A/J mice develop particularly severe myocarditis due to both Th1 (IL-12-induced) and Th2 (IL-4-induced) disease. On the other hand, BALB/c mice, which develop only moderate disease, show less evidence of an IL-4 dependence and Th2 contribution.
A second Th2 cytokine, IL-13, was investigated by Daniela Cihakova (Cihakova and others 2008). She found that IL-13 protects BALB/c mice from myocarditis induced either by cardiac myosin peptide immunization or CB3 viral infection. The severe disease that occurs in the absence of IL-13 is characterized by increased leukocyte infiltration, elevated cardiac myosin autoantibodies, and greater cardiac fibrosis. In fact, many of the IL-13 KO mice developed severe dilated cardiomyopathy with impaired cardiac function and died due to heart failure. The hearts of the IL-13 KO mice had increased levels of the proinflammatory cytokines, IL-1β and IL-18, but not IL-17.
Cytokine Regulation of Autoimmune Disease: Th17 Response
In previous studies we found that IL-12 Rβ1 was essential for the development of autoimmune myocarditis. On the other hand, IFN-γ, the prototypic cytokine of Th1 responses, actually downregulated the severity of disease. We had to consider, therefore, that another cytokine operating through the same receptor may be responsible for the abrogation of disease. At the time of the studies, it became clear that IL-23 also acts on the IL-12 receptor. The 2 cytokines, IL-12 and IL-23, share the same P40 subunit, but IL-12 has a P35 subunit, whereas IL-23 possesses a unique P19 subunit. The following experiments were carried out by a graduate student, Christian Baldeviano. To separate the relative contribution of IL-12 and IL-23 to myocarditis, he immunized BALB/c mice deficient in P40 subunit and found that the dilution completely prevented myocarditis (Baldeviano and others 2010). On the other hand, mice deficient in the P35 subunit were comparable to the wild-type controls. We concluded, therefore, that IL-23 is probably required for autoimmune myocarditis. To make sure that IL-23 is capable of inducing autoimmune myocarditis in P40 KO mice, Christian reconstituted the animals with either IL-12 or IL-23. After immunization with the cardiac myosin peptide in complete Freund adjuvant, we found that IL-23 reconstituted P40 KO mice developed typical cardiac muscle inflammation and necrosis. In contrast, reconstitution with IL-12 resulted in only low-grade disease. Thus, we concluded that IL-23 is sufficient for the development of severe autoimmune myocarditis.
To assess the relative contributions of Th1 and Th17 cells in cardiac inflammation, we quantitated the subpopulation of CD4+ T lymphocytes taken directly from the heart on day 21, the peak time of myocarditis. About equal numbers of IL-17A and IFN-γ producing cells were present in the hearts, indicating that both lineages contributed to the final picture of cardiac inflammation. To estimate how much each subset of T cells contributed to cardiac pathology, Christian used an adaptive transfer model of autoimmune myocarditis. Th1 and Th17 cell lines were generated separating from a CD4+ precursor and stimulated with the myocarditogenic peptide. The cells were then transferred to irradiated syngeneic recipients. Th1 cells produced only mild disease in the recipients, whereas the IL-17-polarized cells induced severe myocarditis with extensive infiltration and disruption of the normal myocardial architecture. In a head-to-head comparison, therefore, IL-17 cells are more pathogenic in myocarditis than IL-1 cells.
The hallmark of TH17 cells is the secretion of cytokines of the IL-17 family, including IL-17A, IL-17F, and IL-22. These cytokines were tested by injecting the respective specific monoclonal antibodies to groups of mice immunized with the myocardidogenic peptide in complete Freund adjuvant (Baldeviano and others 2010). Surprisingly, treatment with antibody to IL-17A produced little or no reduction in the incidence or severity of myocarditis and antibody to IL-17F resulted only in a modest decrease in disease. In contrast, neutralization of IL-22 by monoclonal antibody significantly enhanced the severity of myocarditis when compared with control mice, indicating that this cytokine has a protective role in the disease. We concluded that none of the 3 prototypic Th17-cytokines tested, IL-17A, IL-17F, and IL-22, is essential for the development of the inflammatory phase of autoimmune myocarditis.
To investigate further the role of IL-17A in cardiac inflammation, Christian immunized IL-17A genetically deficient mice with the myocarditogenic peptide in CFA. Although the IL-17A mice developed myocarditis with an incidence and severity similar to controls on day 21, we found that the IL-17A-deficient animals did not develop the severe, life-threatening dilated cardiomyopathy seen in wild-type mice. In fact, IL-17A KO animals were completely protected from the heart failure that lead to the death of many of the controls.
To study the mechanism of protection from dilated cardiomyopathy in IL-17A-deficient mice, we measured cytokines in heart infiltrates after immunization with the myocarditogenic peptide CFA. The most striking finding was a substantial reduction in levels of IL-6 on days 14 and 21 after immunization (Cihakova and Rose 2008). These results suggested, but did not establish, that IL-6 is critical in the progression from inflammatory myocarditis to fibrotic dilated cardiomyopathy. This protection from dilated cardiomyopathy in IL-17A-deficient mice is associated with decreased myocardial fibrosis even as early as 18 days after immunization. Further, we found a significant reduction in matrix metalloproteinase 2 (MMP2) and MMP9, 2 matrix metalloproteins that are associated with cardiac remodeling. Conversely, expression of the tissue inhibitors of the 2 metalloproteinases (TIMP1 and TIMP4) was increased in the IL-17A-deficient animals.
A final series of experiments was carried out to determine if neutralization of IL-17A with monoclonal antibody could be used to prevent the development of dilated cardiomyopathy and the concordant heart failure. We found that treatment with anti-IL-17A monoclonal antibody significantly reduced the interstitial fibrosis and inflammation and reversed the cardinal signs of cardiac failure (Baldeviano and others 2010).
Correlation with Human Heart Failure
Having traced the cytokine pathways from an initial viral infection to autoimmune myocarditis and on to dilated cardiomyopathy, we wondered how relevant these findings are to human heart failure. We were fortunate to collaborate with Dr. Nima Rezaeli and her colleagues in putting together a comprehensive review of the current literature on the role of cytokines in heart failure (Hedayat and others 2010). Briefly, after an initial injury there is increased production of the inflammatory cytokines, IL-1, TNF-α, IL-6, and IL-18, mirroring the effects of infection in mice. Yet, each of these cytokines can act as a double-edged sword. For example, TNF-α acts through 2 receptors, TNF-R1 and TNF-R2. TNF-R1 stimulation by P55 mediates the majority of the injurious effects, whereas TNF-R2 (P75) appears to be responsible for protective effects on the heart. The adverse effects include cardiac myocyte hypertrophy, contractile dysfunction, myocyte apoptosis, and extracellular matrix remodeling.
Similarly, the IL-6-related cytokines can either support or decrease cardiac performance. IL-6 signaling through IL-6 receptor prompts cardiac contractility, but overload leads to maladaptive hypertrophy. IL-6 treatment of cardiac myocytes before induction of ischemia reperfusion injuries associated with decreased mitochondrial depolarization increased mitochondrial Ca2+ loading and decreased cytosolic Ca2+. By interaction with TNF-α and IL-1, it acts to depress cardiac function.
IL-1 has multiple effects on cardiac function. In synergy with TNF-α, IL-1 exacerbates cardiac myocyte and intact heart contraction producing delayed and prolonged myocardial contractility. It also acts by interfering with Ca2+ homeostasis. With TNF-α, it considerably enhances collagen synthesis and induction of MMP2 and MMP9.
IL-18, like IFN-γ, is a proinflammatory cytokine in humans generated during infectious and other inflammatory processes. In synergy with IL-12, it stimulates IFN-γ production by T cells, natural killer cells, and macrophages. IL-18 can also act through IFN-γ-independent mechanisms. It is associated with cardiac myocyte hypertrophy and eventual functional decompensation as well as increased cardiac myocyte apoptosis.
The varying effects of many cytokines, singly or in combination, may explain why so many of the clinical trials using anticytokine therapy in human cardiac inflammatory disease have given rise to ambiguous results. Given questions of timing, dosage, and stage and type of cardiac inflammation, an individual cytokine can play either a physiological or pathological role. Since the cytokines are so interconnected, altering one induces changes in levels of other cytokines. If cytokine treatment is going to be effective in the future, it will require a systematic understanding of the intregative relationships of these mediators, their interactions with receptors and subsequent signaling pathways, and the subsequent effects of unbalancing the cytokine network (Hedayat and others 2010). That is not to say that cytokine-based therapy will not find a place in the treatment of inflammatory heart disease. Quite the contrary, we have only started to realize their potential therapeutic applications. The future depends on garnering sufficient knowledge of their actions and interaction to maximize benefits and reduce risks.
Footnotes
Acknowledgments
This article summarizes the findings of a number of associates whose work is detailed in the references. I thank Starlene Murray for her expert editorial assistance. The original research was supported by PHS grants HL67290, HL70729, and AI151835.
Author Disclosure Statement
No competing financial interests exist.