
Abstract
Interferon alpha (IFN-α) is a critical mediator of human systemic lupus erythematosus (SLE). This review will summarize evidence supporting the role for IFN-α in the initiation of human SLE. IFN-α functions in viral immunity at the interface of innate and adaptive immunity, a position well suited to setting thresholds for autoimmunity. Some individuals treated with IFN-α for chronic viral infections develop de novo SLE, which frequently resolves when IFN-α is withdrawn, supporting the idea that IFN-α was causal. Abnormally high IFN-α levels are clustered within SLE families, suggesting that high serum IFN-α is a heritable risk factor for SLE. Additionally, SLE-risk genetic variants in the IFN-α pathway are gain of function in nature, resulting in either higher circulating IFN-α levels or greater sensitivity to IFN-α signaling in SLE patients. A recent genome-wide association study has identified additional novel genetic loci associated with high serum IFN-α in SLE patients. These data support the idea that genetically determined endogenous elevations in IFN-α predispose to human SLE. It is possible that some of these gain-of-function polymorphisms in the IFN-α pathway are useful in viral defense, and that risk of SLE is a burden we have taken on in the fight to defend ourselves against viral infection.
Introduction
Serum IFN-α Levels Are High in Human SLE
As early as the 1970s, investigators have detected elevated levels of type I IFN in SLE patient sera (Hooks and others 1979), and this observation was confirmed by other groups in the 1980s (Ytterberg and Schnitzer 1982; Kim and others 1987). In the early 2000s, interest in type I IFN in SLE pathogenesis was reinvigorated by a series of interesting results. At this time, the first gene expression microarray studies of peripheral blood cells in SLE were being done, and it was clear that overexpression of type I IFN-induced genes was a common dominant pattern in human SLE (Baechler and others 2003; Bennett and others 2003; Crow and others 2003). This evidence of type I IFN signaling in peripheral blood demonstrated by overexpression of a large number of type I IFN-induced transcripts has been called an IFN signature. In vitro studies in the early 2000s also suggested importance of IFN-α in SLE. Human monocytes exposed to SLE sera could differentiate into activated dendritic cells capable of presenting self-antigens, and this maturation could be inhibited with the addition of anti-IFN-α antibodies (Blanco and others 2001). These experiments suggested the capacity for IFN-α in SLE sera to cause an in vitro tolerance break. Taken together, these results supported the idea that IFN-α was a major cytokine involved in the pathogenesis of human SLE, and stimulated increased investigation in the area.
IFN-α Can Induce SLE in Humans
In addition to the experimental results above, the human experiment of giving recombinant human IFN-α as a therapeutic supports the idea that IFN-α can cause SLE. A number of reports describing IFN-α-induced SLE in patients receiving recombinant human IFN-α to treat malignancy and chronic viral infection have been published (Ronnblom 1990; Ioannou and Isenberg 2000; Niewold and Swedler 2005). The patients described in these reports show the highly specific and characteristic manifestations typical of idiopathic SLE, including malar rash, lupus nephritis, and specific autoantibody responses, including anti-Sm and anti-double-stranded native DNA (dsDNA). When IFN-α was withdrawn, many of these cases improved or resolved, supporting the idea that the IFN-α was causal (Niewold and Swedler 2005). In cohorts treated for chronic hepatitis C, <1% of patients have developed classical de novo SLE (Gota and Calabrese 2003), whereas many more developed an SLE-like syndrome, which was not sufficient to meet formal diagnostic criteria (Ioannou and Isenberg 2000). While not everyone who is given recombinant IFN-α develops SLE, in some individuals exogenous IFN-α is sufficient to induce SLE autoimmunity. This human experience provides a proof of principal that IFN-α can break tolerance (Niewold 2008), and that this IFN-α-induced tolerance break in some individuals results in the very specific autoimmune phenotype of SLE.
High Serum IFN-α Is a Heritable Risk Factor for SLE
SLE runs in families, with first-degree relatives of SLE patients having an approximately 20-fold increased risk of SLE as compared to the general population (Harley and others 2006). Interestingly, other autoimmune diseases are also enriched in SLE families, such as autoimmune thyroid disease (Scofield and others 2007), type I diabetes (Hemminki and others 2009), and rheumatoid arthritis (Alarcon-Segovia and others 2005), and IFN-α has been implicated in the pathogenesis of these disorders to some degree as well (Devendra and Eisenbarth 2004; Mavragani and others 2007; Roelofs and others 2009). Abnormally high levels of IFN-α are present in healthy first-degree relatives of SLE patients as compared to healthy unrelated subjects (Niewold and others 2007, 2008a), suggesting that high serum IFN-α is an inherited risk factor for SLE. It is quite possible that the clustering of high IFN-α in SLE families is related to the clustering of IFN-α-associated disorders in these same families (Niewold and others 2009, 2011). High levels of IFN-α were not observed in spouses of SLE patients, suggesting that genetic and not environmental influences are the cause of this familial clustering. A number of genetic variants have now been associated with increased IFN-α in SLE (Kariuki and others 2008, 2009a, 2009b, 2010a; Kariuki and Niewold, 2010), outlining some of the genetic architecture of this SLE-associated trait and supporting the concept of heritability. The familial nature of the high IFN-α trait was common across SLE patients of all ancestral backgrounds (Niewold and others 2007), suggesting that high serum IFN-α was a common shared pathway to SLE susceptibility.
SLE-Associated Autoantibodies Are Strongly Associated with Serum IFN-α in SLE Patients
SLE patients frequently produce autoantibodies that can bind either dsDNA antibodies or small RNA-binding proteins such as Ro, La, Sm, and RNP. Immune complexes formed by these autoantibodies contain DNA or RNA, respectively. These immune complexes can deliver nucleic acid to the endosomal Toll-like receptors (TLRs) via Fc receptors, and in this way may pathologically activate normal anti-viral immunity. In vitro experiments have supported this idea showing that SLE-associated immune complexes can induce IFN-α production in human peripheral blood mononuclear cell and dendritic cell cultures (Lovgren and others 2004, 2006) (Fig. 1).
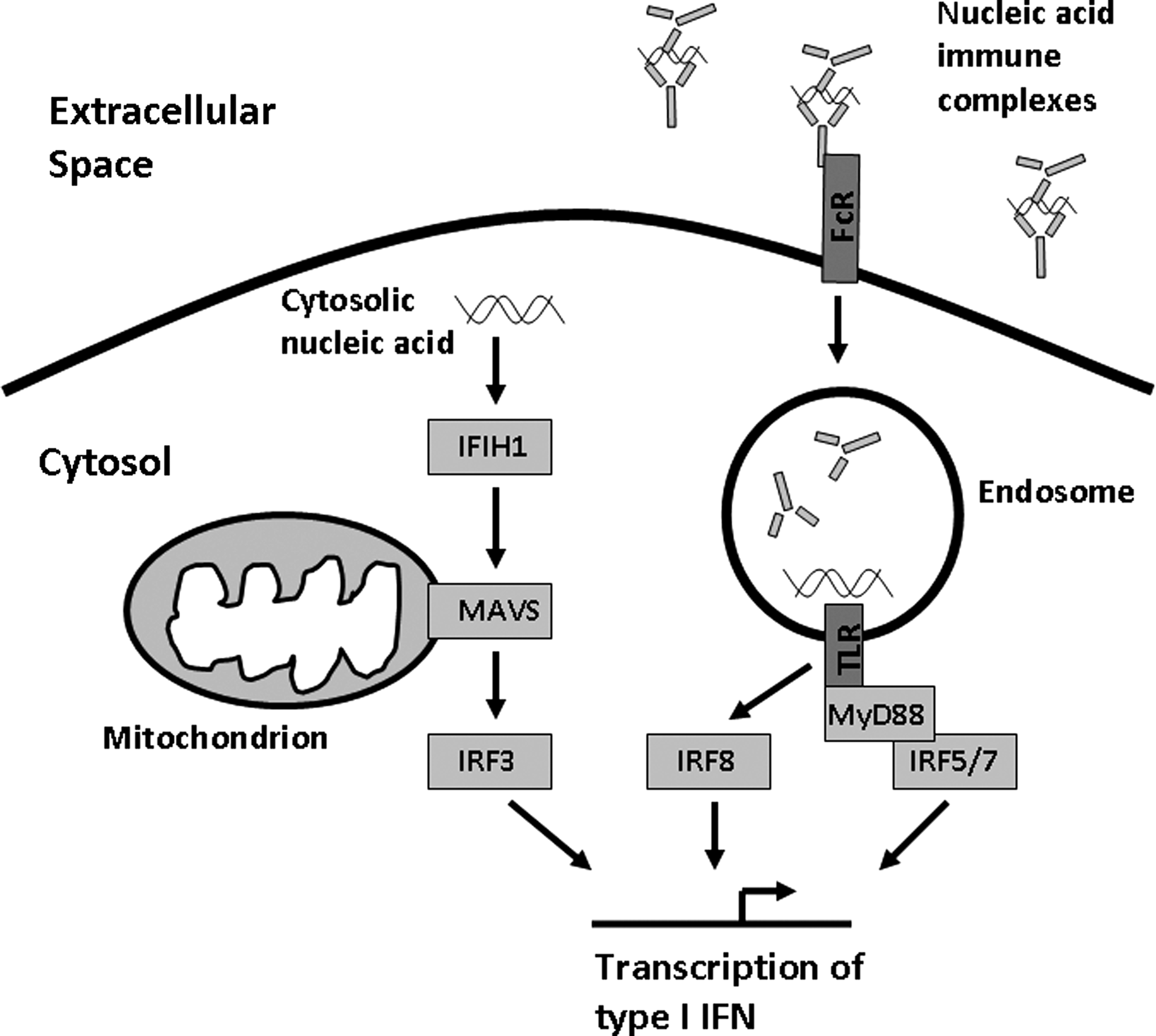
Diagram of the endosomal Toll-like receptor (TLR) and cytosolic nucleic acid sensing pathways of viral defense, showing the location of a number of systemic lupus erythematosus risk loci within these pathways.
In SLE patients in vivo, autoantibody traits provide the strongest association between serum IFN-α and clinical features in SLE, and this association extends to SLE patients of all ancestral backgrounds (Niewold and others 2007; Weckerle and others 2011). While this clinical association is strong and in vitro models suggest a causal relationship between autoantibodies and IFN-α via the endosomal TLR system, the presence of these autoantibodies is not completely predictive of high IFN-α in patients in vivo (Niewold and others 2008c). This suggests that other host factors influence the relationship between autoantibodies and serum IFN-α in humans. A two-hit model may apply, in which the formation of SLE-associated autoantibodies exacerbates an underlying genetic tendency toward greater IFN-α production or greater IFN-α sensitivity, resulting in the clinical development of SLE (Niewold and others 2007; Niewold and others 2010). It is known that SLE-associated autoantibodies can be found in human sera years before the development of clinical SLE (Arbuckle and others 2003), and it is possible that this preclinical period is characterized by amplification of serum IFN-α as suggested in this two-hit model.
SLE-Associated Genetic Variations in the IFN-α Pathway Are Gain of Function
A number of the genetic polymorphisms that confer susceptibility to SLE are found in genes that function in the IFN-α pathway. Given the data summarized above indicating that high IFN-α is a causal heritable trait in human lupus, it seems likely that IFN-α pathway polymorphisms associated with SLE would be gain of function in nature (Kariuki and Niewold 2010). Polymorphisms that have demonstrated an effect upon the IFN-α pathway in SLE patients in vivo are summarized in Table 1. Genetic variations in both IRF5 and IRF7 have been associated with SLE (Sigurdsson and others 2005; Graham and others 2006; Harley and others 2008). These transcription factors function downstream of the endosomal TLRs, and can induce transcription of IFN-α. Interestingly, the genetic variations in IRF5 and IRF7 were linked to higher IFN-α in SLE patients, but only in those patients who had particular SLE-associated autoantibodies (Niewold and others 2008b; Salloum and others 2010). This suggested a “gene + autoantibody=high IFN-α” model, in which the autoantibodies presumably act like an endogeneous TLR stimulus that brings out the genetic effect upon serum IFN-α at these loci (Salloum and Niewold 2011). The polymorphism in the osteopontin gene (SPP1) that has been associated with SLE was associated with higher serum osteopontin and higher IFN-α (Kariuki and others 2009b). Interestingly, there was an age- and sex-related influence, with males and younger female SLE patients demonstrating a large genetic effect upon cytokine levels that was not shared with older female SLE patients (Kariuki et al. 2009b; Weckerle and Niewold 2011). The polymorphism in PTPN22 that has been linked to SLE as well as other autoimmune diseases was associated with increased serum IFN-α and decreased serum TNF-α in the same serum sample (Kariuki and others 2008). IFN-α has been implicated in the pathogenesis of some of the other autoimmune diseases associated with PTPN22, including autoimmune thyroid disease, type I diabetes, and rheumatoid arthritis (Devendra and Eisenbarth 2004; Mavragani and others 2007; Roelofs and others 2009).
IFN, interferon alpha; SLE, systemic lupus erythematosus.
While variations within the IFN-α gene locus have not been associated with SLE susceptibility to date, a recent study reported evidence for association of the IFN-κ gene with SLE (Harley and others 2010). SNPs in the IFNK locus were also associated with serum IFN-α (Harley and others 2010). IFN-α is the major circulating type I IFN in SLE sera (Niewold and others 2007), whereas IFN-κ is constitutively expressed in the skin (LaFleur and others 2001). It is possible that IFN-κ in the skin may prime resident plasmacytoid dendritic cells to produce more IFN-α, which is then detectable in the serum. While each of the above polymorphisms has been linked to greater serum IFN-α activity, other potential mechanisms for increasing pathway signaling are also possible. The STAT4 risk allele that has been associated with risk of both SLE and rheumatoid arthritis (Remmers and others 2007) was not associated with higher serum IFN-α in SLE patients. Instead, this polymorphism was associated with increased IFN-α-induced gene expression for a given amount of IFN-α (Kariuki and others 2009a). These data suggest that the autoimmune disease-associated polymorphism in STAT4 increases the cellular sensitivity to IFN-α, modulating the pathway downstream of the type I IFN receptor.
Loss-of-Function Polymorphism in the TLR-Independent Pathway of Viral Defense Leads to Lower IFN-α in SLE Patients
It is likely that the TLR-independent pathway of viral response will be involved in SLE pathogenesis as well. Genetic variation in the cytosolic RNA sensor IFIH1 (MDA5) has been associated with human SLE (Harley and others 2008; Gateva and others 2009), and the p200 family of proteins that can sense cytosolic DNA are important in murine SLE models (Choubey and others 2010). MAVS is an adaptor protein located in the mitochondrion that mediates signaling of the IFIH1 and RIG-I cytosolic nucleic acid sensors (Fig. 1). We studied reported human genetic variants in MAVS that resulted in coding changes, and identified one critical loss-of-function coding change variant that resulted in a C79F amino acid substitution (Pothlichet and others 2011). This polymorphism was associated with a large decrease in type I IFN and inflammatory cytokine production in human cell lines (Pothlichet and others 2011). In human SLE patients, this coding change polymorphism was associated with decreased serum IFN-α, and was enriched within this group of patients (Pothlichet and others 2011). This study reinforces the concept of genetic influence upon IFN-α responses, and reminds us that not all SLE patients have high IFN-α, and likely the pathogenic factors relevant to the low IFN-α group will differ from the high IFN-α group.
Novel Genetic Loci Associated with IFN-α and Autoantibodies in SLE
Given the strong association of many known SLE genetic risk factors with alterations in serum IFN-α, we hypothesized that the serum IFN-α trait could be used to discover novel genes that were important to SLE pathogenesis. Because the IFN-α trait is causal in human SLE, genes related to this trait should be associated with the early pathogenic events in SLE. By studying a quantitative intermediate trait, the power of the study is greatly increased. We compared genetic markers across the genome in a genome-wide study of SLE patients who were in the highest third of serum IFN-α with those in the lowest third of IFN-α to maximize the difference between the groups (Kariuki and others 2010a). Because autoantibodies are so closely related to serum IFN-α in SLE, patients were also stratified by the presence or absence of the SLE-associated autoantibodies anti-Ro, anti-La, anti-Sm, anti-RNP, and anti-dsDNA. Thus, polymorphisms that were associated with IFN-α and/or autoantibodies could be detected using this design. Eight novel loci were detected in this study (Kariuki and others 2010a, 2010b), and these all demonstrated some association with serum IFN-α (Table 2). While some genetic polymorphisms demonstrated a direct relationship with serum IFN-α (LRRC20 and PTPRM), more frequently the genetic variation was associated with a particular autoantibody profile, and then this autoantibody profile was associated with higher IFN-α (this pattern of association is labeled as “secondary” in Table 2). Many of these gene loci that were associated with IFN-α and autoantibodies in SLE patients have not been previously studied extensively, although many have likely functions within the immune system. A summary of the known functional data regarding each gene encoded by the associated locus is provided in Table 2.
dsDNA, double-stranded native DNA; TLR, Toll-like receptor.
Conclusions
In summary, multiple lines of evidence support the involvement of IFN-α in the primary pathogenesis of human SLE. While exogenous IFN-α administration does not initiate SLE in all subjects, we have found intriguing genetic evidence that signaling through this pathway is highly polymorphic in humans, and this may explain some of this heterogeneity in response to exogenous IFN-α. This same heterogeneity in IFN-α signaling appears to underlie idiopathic SLE as well. While many of the gain-of-function polymorphisms in the IFN-α pathway could have beneficial functions in viral defense, it is possible that some individuals inherit too much of a good thing, resulting in significant risk of SLE. Given the high burden of genetic selection related to infectious disease throughout human history, this scenario seems likely, and we as humans have probably been specifically enriched for some of these autoimmune disease risk alleles. Novel therapeutics targeting the IFN-α pathway are currently in clinical trials for SLE. Variability in the response to these agents will likely relate to some of the genetic variations we have outlined above.
Footnotes
Acknowledgments
This work was supported by NIH K08 AI083790, NIH P30 DK42086, NIAID Clinical Research Loan Repayment AI071651, NIH CTSA Core Subsidy Grant, and CTSA Pilot Grants from UL1 RR024999, Lupus Research Institute Novel Research Grant, Alliance for Lupus Research Target Identification in Lupus Grant.
Author Disclosure Statement
No financial conflict of interest to declare.