
Abstract
The nuclear factor-kappa B (NFκB) signal transduction pathway plays an important role in immunity, inflammation, cell growth, and survival. Since dysregulation of this pathway results in high, constitutive NFκB activation in various cancers and immune disorders, the development of specific drugs to target this pathway has become a focus for treating these diseases. NFκB regulates various aspects of the cellular response to interferon (IFN). However, the role of the upstream regulator of the NFκB signaling pathway, the inhibitor of κB kinase (IKK) complex, on IFN function has not been examined. In the present study, we examined the effects of 2 IKK inhibitors, N-(1,8-Dimethylimidazo[1,2-a]quinoxalin-4-yl)-1,2-ethanediamine hydrochloride (BMS-345541) and 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1), on IFN action in several human glioma cell lines. IKK inhibitors inhibit glioma cell proliferation, as well as TNF-induced RelA (p65) nuclear translocation and NFκB-dependent IL8 gene expression. Importantly, BMS-345541 and TPCA-1 differentially inhibit IFN-induced gene expression, completely suppressing MX1 and GBP1 gene expression, while having only a minor effect on ISG15 expression. Furthermore, these IKK inhibitors displayed marked differences in blocking IFN-induced antiviral action against cytopathic effects and replication of vesicular stomatitis virus (VSV) and encephalomyocarditis virus (EMCV). Our results show that the IKK complex plays an important function in IFN-induced gene expression and antiviral activity. Since VSV and EMCV are oncolytic viruses used in cancer therapy, our results indicate the potential synergy in combining IKK inhibitors with oncolytic viruses.
Introduction
Interferons (IFNs) are a family of endogenously expressed glycoproteins initially discovered because of their potent antiviral activity. IFNs also affect cell proliferation, differentiation, and survival; angiogenesis; and the immune system (Pfeffer and others 1998). Because of these diverse actions, IFNs have proven to be useful clinically in a variety of diseases of diverse pathogenesis and phenotypes, including various forms of cancer (Belardelli and others 2002). IFNs are currently among the most commonly used agents employed in the biological therapy of cancer. The antitumor efficacy of IFN is based on its multiple effects. Upon binding to IFN receptors, IFN induces the activation of receptor-bound tyrosine kinases JAK1 and TYK2, which subsequently phosphorylate the signal transducer and activator of transcription (STAT) factors, resulting in their nuclear translocation where they bind to IFN-stimulated gene (ISG) promoters and induce ISG expression (Darnell and others 1994).
We previously showed that the NFκB transcription factor suppresses various aspects of the cellular response to IFN (Yang and others 2000; Yang and others 2001; Yang and others 2010). Moreover, not only is NFκB constitutively active in a variety of cancers, but IFN can also promote NFκB activity (Du and others 2007). IFNs have also been previously employed in the treatment of glioma (Lundblad and Lundgren 1981; Nederman and others 1981; Miki 1982). Although the IKK complex plays an important role in the innate response via the pattern recognition pathway upon viral and bacterial infections leading to IFN production (Hacker and Karin 2006; Meylan and others 2006), it is unresolved whether the IKK complex is also directly involved in the IFN-mediated antiviral signal transduction pathway. In the present study, we examined the effect of IKK inhibitors on the IFN response in human glioma cells. Our results showed that the IKK inhibitors, N-(1,8-Dimethylimidazo[1,2-a]quinoxalin-4-yl)-1,2-ethanediamine hydrochloride (BMS-345541) and 2-[aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1), suppress NFκB activity, differentially attenuate the expression of ISGs, and markedly inhibit the IFN-induced antiviral activity. These results strongly suggest that IKK complexes directly regulate the IFN-mediated signal transduction pathway and a specific subset of ISGs. Our findings also opened a new avenue to enhance the efficacy of oncolytic viruses to treat various cancers by using these IKK inhibitors.
Materials and Methods
Biological reagents and cell cultures
Recombinant human IFNα (IFNCon1) was generously provided by InterMune. The biological activity of the IFN preparations was expressed in terms of international reference units/ml using the appropriate National Institutes of Health reference standard (Pfeffer and others 1997). Recombinant human tumor necrosis factor α (TNF-α) was purchased from R&D Systems, Inc. Antibodies against the following proteins were used: p65, STAT1, STAT2, and actin (Santa Cruz Biotechnology); phospho-STAT1 (Cell Signaling Technology); and phospho-STAT2 (Millipore). U87, MT330, SJ-G2, and GBM6 cells were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (Hyclone Laboratories). Cells were maintained in the presence of penicillin (100 IU/mL) and streptomycin (100 ìg/mL) at 37°C with 5% CO2. BMS-345541 and TPCA-1 were obtained from Sigma-Aldrich and dissolved in dimethyl sulfoxide.
Cell viability assay
Ten microliters of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) from stock solution (10 mg/mL) was added to each well of 96-well plates containing glioma cells and incubated at 37°C for 2–4 h. Oxidized MTT was solubilized by adding 100 μL of 10% sodium dodecyl sulfate (SDS) in 0.01 N HCL, and plates were incubated at 37°C for 4 h in a humidified chamber. Plates were read at 570 nm on a Bio-Rad plate reader.
Antiviral assays
Cells at 50%–80% confluence in 6-well plates were treated with various concentrations of BMS-345541, TPCA-1, or IFNα, either separately or in combination for 24 h, followed by infection with vesicular stomatitis virus (VSV) or encephalomyocarditis virus (EMCV) for 1.5 h at a multiplicity of infection of ∼0.1 plaque-forming unit per cell. The VSV yield in the medium was assayed by plaque formation at 24 h postinfection on Vero cells (Yang and others 2000). The EMCV yield in the medium was assayed by quantitative real-time reverse transcriptase–polymerase chain reaction (qRT-PCR) using an EMCV 3D gene-specific primer pair: 5′-CCCTACCTCACGGAATGGGGCAAA-3′ (forward), 5′-GGTGAGAGCAAGCCTCGCAAAGAC-3′ (reverse) (Perez and Diaz de Arce 2009). Viral RNA from 200 μL of medium was isolated using TRIZol reagent (Invitrogen). Data were normalized to the viral RNA sample isolated from EMCV stock with known viral titer.
Immunofluorescence staining
Cells were grown in 48-well plates, pretreated with BMS-345541 or TPCA-1 for 2 h, and stimulated with recombinant TNF-α for 30 min or IFN for 1 h. Cells were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton×100. After blocking with 5% goat serum, cells were incubated with anti-p65, anti-STAT2, or anti-pSTAT1, and subsequently stained with Alex 594-labeled goat anti-rabbit IgG (Invitrogen).
Preparation of total cell lysates and western immunoblot
Cells were washed twice with ice-cold PBS, and lysates were prepared by incubation in lysis buffer (30 min, 4°C) containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.5% Nonidet P-40%, 15% glycerol, 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethanesulfonylfluoride or phenylmethylsulfonyl fluoride (PMSF), 5 μg/mL soybean trypsin inhibitor, 5 μg/mL leupeptin, and 1.75 μg/mL benzamidine (Yang and others 2011). Whole-cell lysates were precleared by centrifugation (12,000 g, 15 min). Total cell lysates (25 μg) were separated by SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes (Millipore), and immunoblotted with the indicated antibodies, followed by IRDye800CW goat anti-mouse IgG or IRDye680 goat anti-rabbit IgG (LI-COR Biosciences). Blots were visualized on an Odyssey Infrared Imaging System (LI-COR Biosciences).
ISG expression by qRT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen), and qRT-PCR was performed on an iCyclerIQ (Bio-Rad) using the iScript One-Step RT-PCR Kit with SYBR Green (Bio-Rad). Reaction parameters were as follows: cDNA synthesis at 50°C for 20 min, transcriptase inactivation at 95°C for 5 min, and PCR cycling at 95°C for 10 sec and 60°C for 30 sec for 40 cycles. The following primers were used for qRT-PCR: β-actin, 5′-AGAAGGAGATCACTGCCCTG-3′ (forward), 5′-CACATCTGCTGGAAGGTGGA-3′ (reverse); ISG15, 5′-TCCTGGTGAGGAATAACAAGGG-3′ (forward), 5′- GTCAGCCAGAACAGGTCGTC-3′ (reverse); GBP1, 5′-AGGAGTTCCTTCAAAGATGTGGA-3′ (forward), 5′- TTCTGAACAAAGAGACGATAGCC-3′ (reverse); MX1, 5′-TCCCACCCTCTATTACTGAATGG-3′(forward), 5′- GGGAAGGGCAACTCCTGAC-3′ (reverse), IL8, 5′-TAGCAAAATTGAGGCCAAGG-3′ (forward), 5′-AGCAGACTAGGGTTGCCAGA-3′ (reverse).
Data analysis
For statistical analysis of the data a minimum of two independent experiments was performed at least in duplicate, and analysis was performed by Student's t-test.
Results
BMS-345541 and TPCA-1 inhibit NFκB activity as well as glioma cell proliferation and IL8 expression
The NFκB signal transduction pathway plays a crucial role in promoting cell survival by enhancing the expression of antiapoptotic genes, as well as suppressing the expression of pro-apoptotic genes (Ben-Neriah and Karin 2011). The constitutively high NFκB activity in various forms of cancer promotes tumor cell survival, which is believed to contribute to inherent resistance to chemotherapy and radiotherapy. We have previously established that NFκB promotes cell survival and regulates the expression of a subset of IFN target genes that are likely effectors of IFN's biological actions (Du and others 2007). Since the IKK complex lies upstream of both the classical and alternative NFκB signaling pathways (Hacker and Karin 2006), we used specific inhibitors of the IKK complex (Burke and others 2003; Podolin and others 2005) to study the role of this complex in IFN action. In the initial series of experiments, we examined the effect of the IKK inhibitor BMS-345541 on the proliferation of human glioma cells. In brief, U87, MT330, SJ-G2, and GBM6 human glioma lines were treated with various concentrations of BMS-345541, and after 3 days viable cells were quantified by MTT staining. As shown in Fig. 1A, BMS-345541, at concentrations of 10 μM and higher, markedly blocked the proliferation (80%–95% inhibition) of U87, MT330, SJ-G2, and GBM6 glioma cell lines. At concentrations between 1 and 10 μM, BMS-345541 had an inhibitory effect on cell proliferation, although the effect was cell type dependent. The sensitivity of the different cells to BMS-345541 was SJ-G2>U87>MT330>GBM6. The dose–response relationship of BMS-345541 on glioma cell proliferation is similar to its reported inhibitory effects on the growth of melanoma cells in vitro and in vivo (Yang and others 2006).

BMS-345541 and TPCA-1 inhibit glioma cell proliferation, NFκB activation, and NFκB-regulated IL8 gene expression:
To determine whether the inhibitory effect of BMS-345541 on cell proliferation was related to inhibition of NFκB activity, U87 cells were pretreated with BMS-345541 for 2 h and then stimulated with TNF-α for 30 min, and immunofluorescence staining was used to monitor the nuclear translocation of RelA/p65 subunit of NFκB. As shown in Fig. 1B, in unstimulated U87 cells, p65 is localized mainly in the cytoplasm, but p65 is translocated into the nucleus upon TNF-α stimulation. Pretreatment of U87 cells with BMS-345541 significantly inhibited p65 nuclear translocation at 5 μM and nearly completely blocked nuclear translocation at 10 μM. Similar results were also obtained with another IKK inhibitor, TPCA-1, at concentrations of 2.5 and 5 μM, which blocked the TNF-induced p65 translocation into the nucleus of U87 cells (Fig. 1B).
To further characterize the effects of both BMS-345541 and TPCA-1 on NFκB-dependent events in glioma cells, we examined the effect of BMS-345541 and TPCA-1 on the TNF-induced expression of the NFκB-regulated gene, IL8. In brief, U87 cells were pretreated (2 h) with various concentrations of BMS-345541 or TPCA-1, and then total RNA was prepared from cells stimulated with TNF for 6 h. IL8 gene expression was determined by qRT-PCR and normalized to the expression of the housekeeping gene β-actin. As shown in Fig. 1C, TNF induced a dramatic increase in IL8 expression. BMS-345541 inhibited IL8 expression in a dose-dependent manner with an IC50 of >2 μM and complete inhibition at >5 μM. BMS-345541 has a reported IC50 of 0.3 μM for IKKβ and 4 μM for IKKα, respectively (Burke and others 2003), when kinase activity was measured in vitro using GST-tagged IκBα as substrate. However, BMS-345541 was only observed to have marked inhibitory effects on cell proliferation and NFκB activity at concentrations ≥10 μM, suggesting that the effects of BMS-345541 involve the inhibition of both IKKβ and IKKα. In Fig.1C, we also examined the effect of TPCA-1 on the expression of IL8. At>1 μM, TPCA-1 almost completely suppressed TNF-induced IL8 expression, which correlates with the concentration needed for the inhibition of p65 translocation.
IKK inhibitors block IFN-induced gene expression in glioma cells
In previous studies, we have shown that NFκB plays an important role in regulating the expression of a subset of IFN-induced genes (Wei and others 2006). Since the data in Fig. 1 demonstrate that IKK inhibitors inhibit NFκB activity in glioma cells, we next examined the effect of BMS-345541 and TPCA-1 on IFN-induced gene expression. U87 and MT330 cells were pretreated with 5 μM BMS-345541 for 2 h and then incubated with IFN (1000 U/mL) for an additional 5 h. Total RNA was prepared, and the level of expression of the IFN-induced genes, ISG15, GBP1, and MX1, was measured by qRT-PCR. As shown in Fig. 2A and B, IFN markedly induced the gene expression of ISG15, GBP1, and MX1 in U87 and MT330 cells. BMS-345541 nearly completely blocked IFN-induced expression of GBP1 and MX1, but only had a slight effect on ISG15 gene expression. Similar effects of BMS-345541 on IFN-induced gene expression were found with GBM6 and SGJ2 glioma cell lines (data not shown). These results indicate that the expression of GBP1 and MX1 are IKK-regulated IFN-induced genes, while ISG15 expression is independent of the IKK pathway. Similar results were also obtained with TPCA-1 as shown in Fig. 2C. TPCA-1 only marginally inhibited the expression of ISG15 at 1 μM, but almost completely attenuated the expression of MX1.
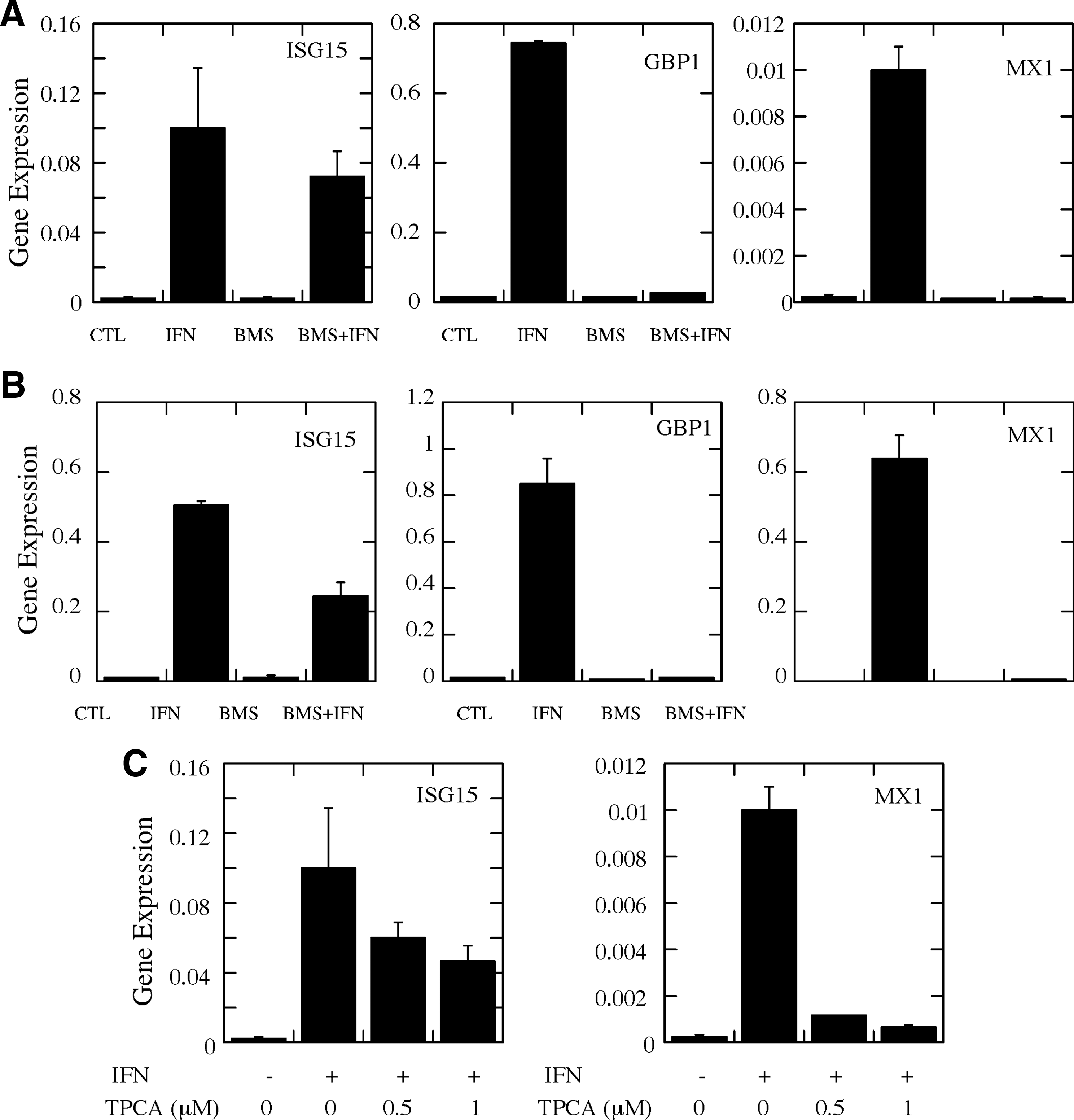
BMS-345541 and TPCA-1 attenuate IFN-induced ISG expression.
Type I IFN-induced expression of ISGs involves activation of the Janus kinase (JAK)–STAT signaling pathway. Since we observed that BMS-345541 treatment inhibited the expression of several IFN-induced genes, we next examined the effect of BMS-345541 on the IFN-induced activation of STAT1 and STAT2. Cells were pretreated with 5 μM BMS-345541 and stimulated with IFN for 1 h, and whole-cell lysates were prepared and immunoblotted for STAT1/STAT2 phosphorylation. As shown in Fig. 3A, BMS-345541 did not affect the IFN-induced activation of STAT1 or STAT2 as evidenced by the detection of phospho-STAT1 and STAT2, respectively. In addition, immunoblotting for STAT1 and STAT2 showed that IFN and BMS-345541 had no effect on the levels of the STAT proteins. Moreover, IFN-induced nuclear translocation of STAT1 and STAT2 in glioma cells (Fig. 3B) was also unaffected by pretreatment of cells with BMS-345541. These results clearly show that, while BMS-345541 has no effect on the JAK-STAT signaling pathway, it specifically blocks the IKK/NFκB signaling pathway.
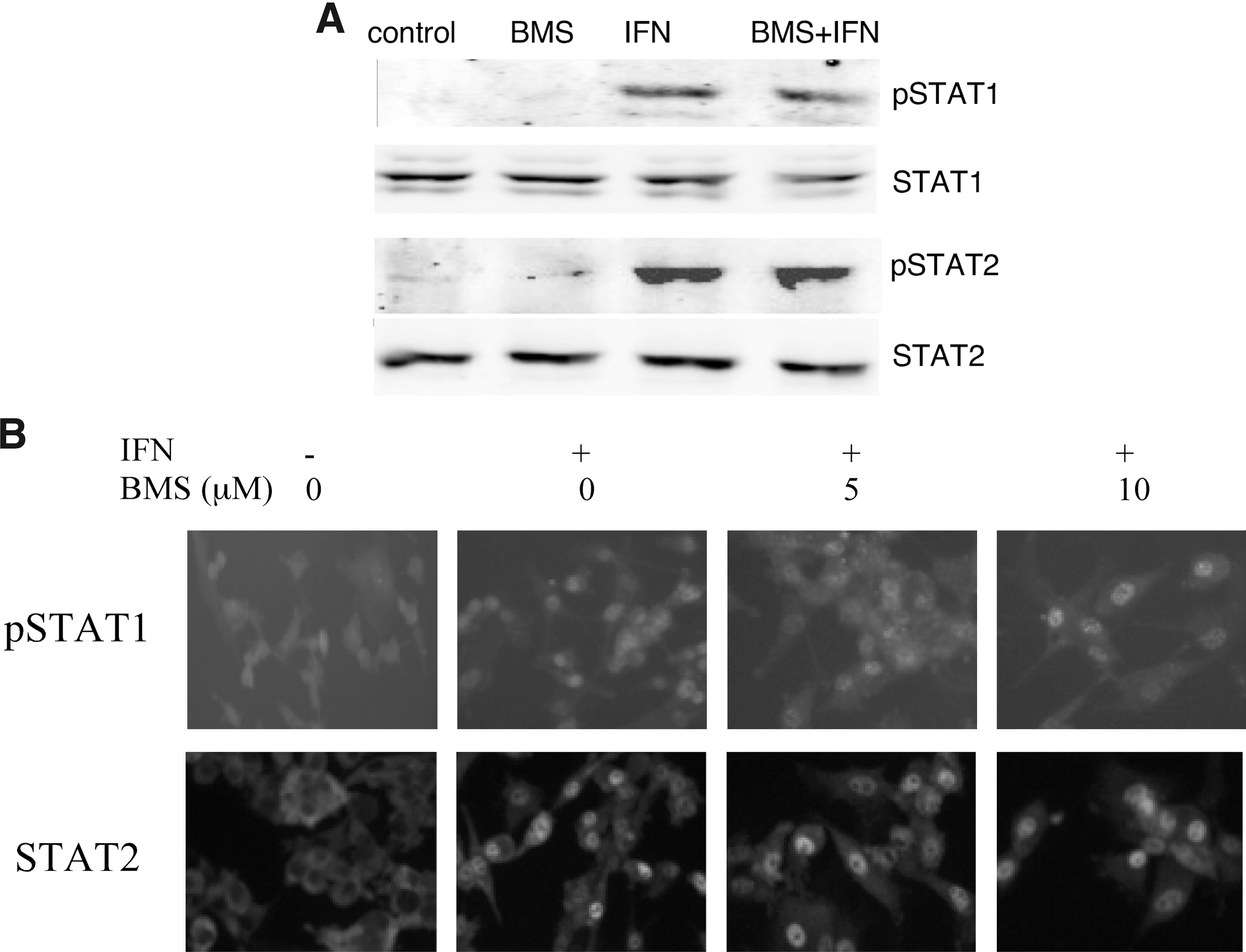
BMS-345541 does not affect STAT1 and STAT2 activation.
BMS-345541 blocks IFN-induced protection against the cytopathic effects of the oncolytic viruses VSV and EMCV
Upon virus infection, cellular sensors (pattern recognition receptors) recognize viral-pathogen-associated molecular patterns and initiate an innate immune response that consequently results in vibrant NFκB activation. NFκB regulates the expression of not only various cytokines, including IFNs, that protect the surrounding cells from further infection, but also genes that are important for cell survival. Furthermore, several viruses have evolved strategies that utilize the NFκB pathway to enhance their replication. Since we found that BMS-345541 selectively inhibited the expression of IFN-induced genes, such as GBP1 and MX1, which play important roles in the antiviral action of IFN, we next examined the effect of BMS-345541 on the ability of IFN to protect glioma cells against virus-induced cytopathicity. In these experiments, glioma cells were pretreated with BMS-345541 before IFN exposure, infected with VSV for 24 h, and then cell death was determined by MTT assays. IFN treatment resulted in varying degrees of protection against VSV-induced cell death, depending on the glioma cell type (Fig. 4A). The differential sensitivity of glioma lines to the antiviral effects of IFN against VSV has been reported previously (Wollmann and others 2007). Most interestingly, at relatively high concentrations (8–10 μM) BMS-345541 was found to inhibit the protective effect of IFN against VSV infection. As shown in Fig 4B, IFN protected U87 cells from virus-induced cell rounding, which is an early event in a VSV-induced cytopathicity. BMS-345541 pretreatment (5 μM) inhibited the protective effect of IFN against VSV-induced cell rounding (Fig. 4B).
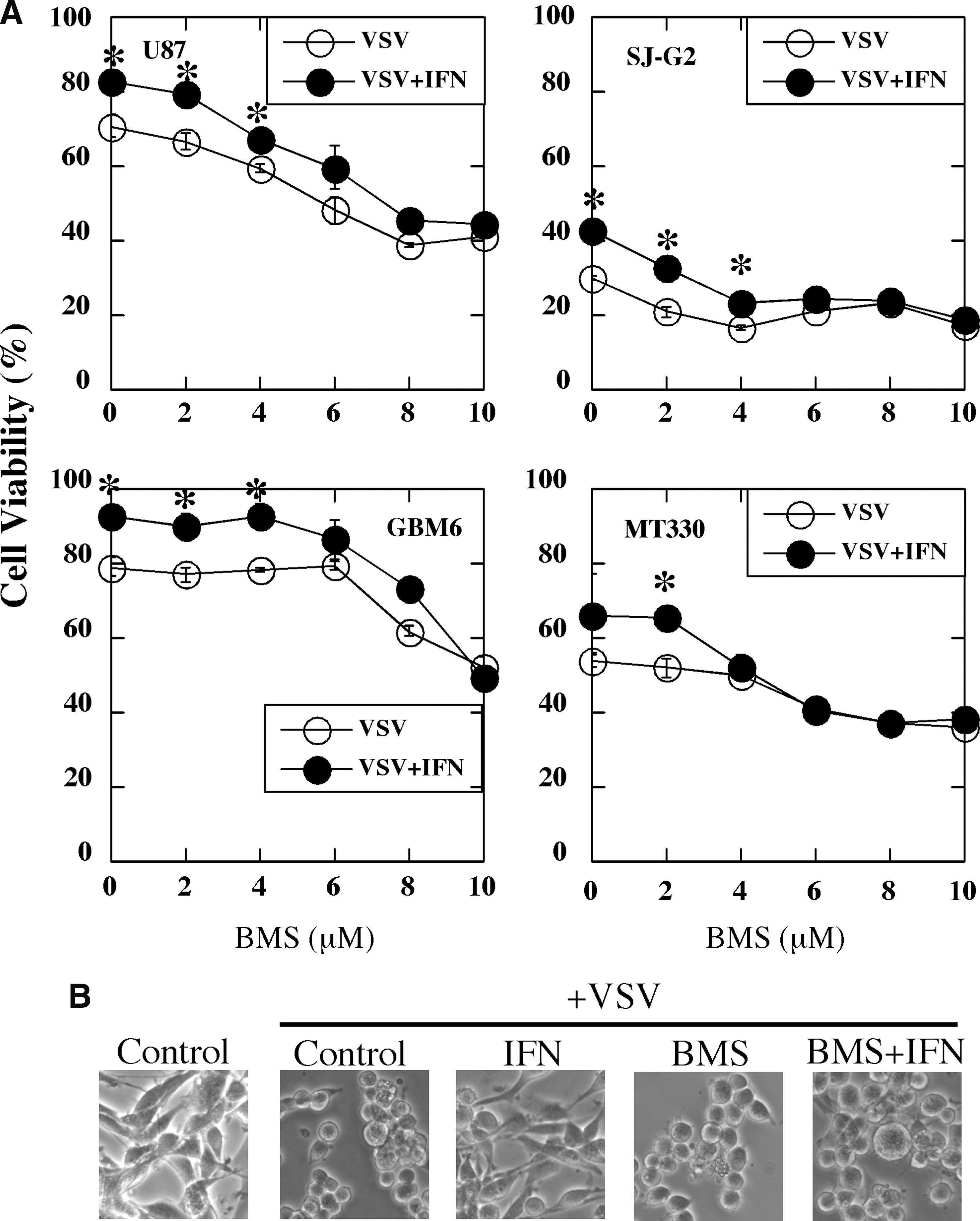
BMS-345541 blocks IFN protection against VSV-induced cytopathic effect in glioma cells.
We next examined the effect of BMS-345541 on the ability of IFN to protect glioma cells against the cytopathic activity of EMCV. As shown in Fig. 5A, IFN treatment was highly effective in protecting glioma cells, especially U87 and SJ-G2 cells, from EMCV-induced cell death, but IFN protection was nearly completely reversed by pretreatment of glioma cells with BMS-345541 at concentrations ≥6 μM. Taken together these results showed that BMS-345541 suppressed the protective action of IFN against VSV- and EMCV-induced cytopathicity and most importantly correlated with the inhibition of NFκB activity by BMS-345541. Moreover, the level of protection induced by IFN in the 4 cell types markedly differed between the 2 viruses.
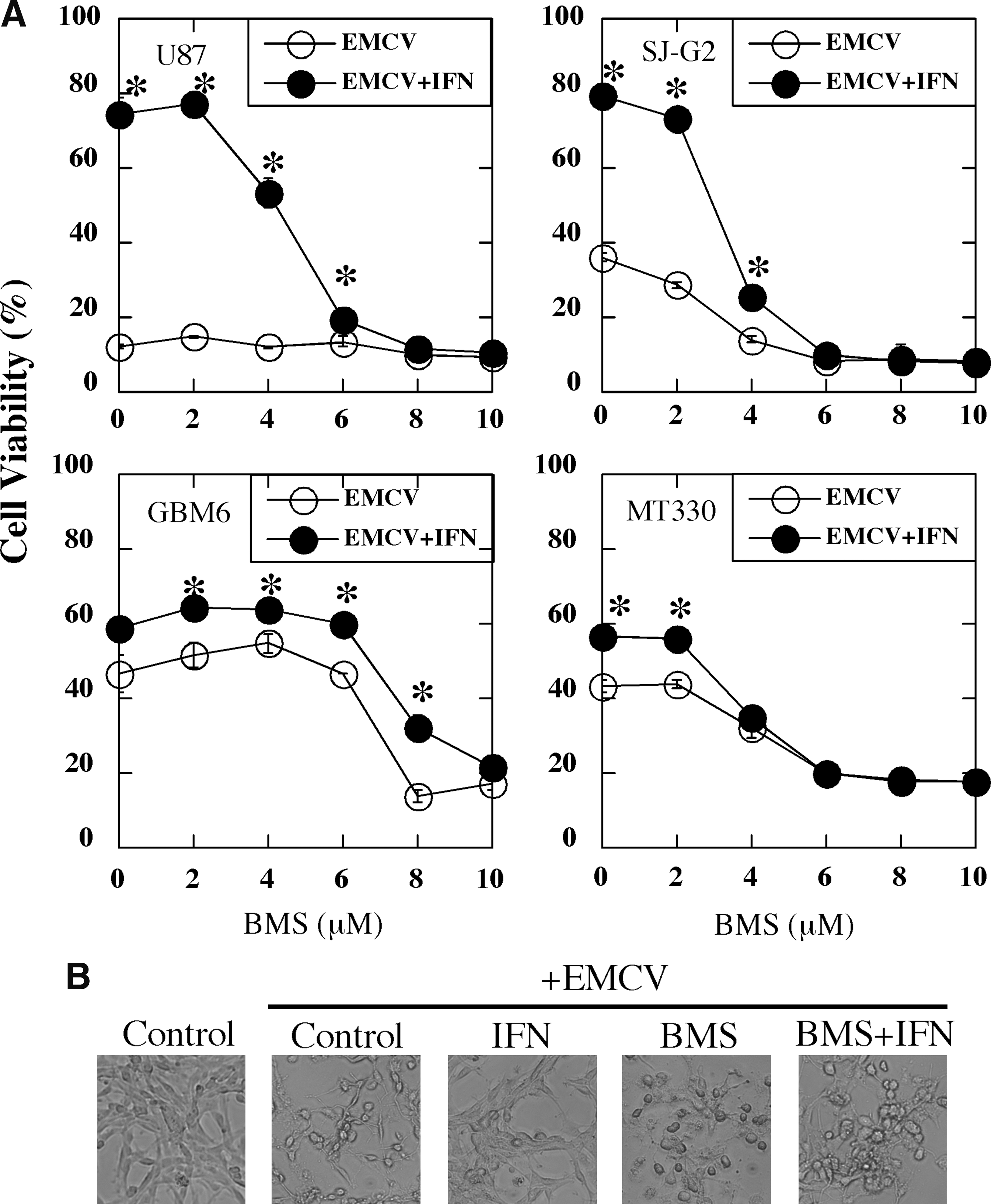
BMS-345541 blocks IFN protection against EMCV-induced cytopathic effect in glioma cells.
BMS-345541 and TPCA-1 suppress IFN-induced antiviral activity against VSV and EMCV in glioma cells
Since the IKK inhibitor BMS-345541 suppressed the ability of IFN to protect glioma cells against the cytopathic effects of VSV and EMCV, we next determined whether BMS-345541 and TPCA-1 could block the ability of IFN to inhibit virus replication in U87 cells. U87 cells were pretreated with BMS-345541 or TPCA-1 before IFN exposure, infected with VSV, and virus replication was assayed by plaque reduction assays. As shown in Fig. 6A, IFN treatment (1000 U/mL) of U87 cells reduced VSV titer by almost 1000-fold. However, pretreatment of U87 cells with 5 μM BMS-345541 or 5 μM TPCA-1 before IFN exposure resulted in a <10-fold reduction in VSV titer, or a >100-fold reduction in IFN antiviral activity. BMS-345541 alone had no effect on VSV replication. Thus, both BMS-345541 and TPCA-1 markedly impaired the ability of IFN to inhibit VSV replication.

BMS-345541 and TPCA-1 attenuate IFN-induced antiviral state against VSV and EMCV in Glioma cells. U87 cells were pretreated with 5 μM BMS-345541 or 5 mM TPCA-1 for 2 h before the addition of IFN (1000 U/mL or varying IFN concentrations for 24 h), and infected with VSV or EMCV. Viral titers were measured at 24 h postinfection by plaque assays for VSV
We next examined the effect of BMS-345541 and TPCA-1 on IFN-induced antiviral activity against EMCV (Fig. 6B). Treatment of U87 cells with IFN concentrations as low as 10 U/mL reduced EMCV titer by ∼1000-fold, and at 1000 U/mL by 10,000-fold. However, the anti-EMCV action of IFN in U87 cells was significantly inhibited by pretreatment of U87 cells with 5 μM BMS-345541, resulting in only a 10-fold reduction in EMCV titer in cells treated with 1000 IU/mL of IFN. BMS-345541 alone did not have any effect on EMCV replication. Similar to the effect of BMS-345541, pretreatment of U87 cells with 5 μM TPCA-1 before IFN treatment resulted in a <10-fold reduction in EMCV titer or >1000-fold reduction in IFN antiviral activity. Our results clearly demonstrate that IKK/NFκB plays very important role in IFN-mediated antiviral activities. IFN showed a very weak anti-VSV activity in U87 cells, reducing VSV titer by ∼1000-fold at 1000 U/mL. However, IFN exhibited marked anti-EMCV activity decreasing EMCV titer by >1000-fold even at 10 U/mL. This suggests that IFN-induced antiviral activities are highly virus dependent. Combining with the results from BMS-345541, these results clearly suggest that there is a crosstalk between the IKK-NFκB pathway and the IFN signal transduction pathway.
Discussion
In this report, we have demonstrated that the IKK complex that lies upstream of NFκB activation plays an important role in regulating several activities of type I IFN. Both IKK inhibitors BMS-345541 and TPCA-1 block the IFN-induced expression of a subgroup of ISGs as well as IFN-induced antiviral activity. Both inhibitors also block NFκB activation induced by the potent NFκB activator TNF but had no effect on the IFN-induced activation of STAT1 and STAT2, demonstrating the high specificity of these IKK inhibitors.
NFκB has been reported to regulate the expression of IFN-induced genes in various cells; however, the effects are cell type dependent (Du and others 2007). For example, the p65 subunit of NFκB also binds to some ISG promoters upon IFN treatment (Wei and others 2006; Yang and others 2007). In addition, we have previously shown that IFN stimulates NFκB activity in various cell lines, and that IFN-induced NFκB activation counteracts the antiproliferative, antiviral, and proapoptotic activities of IFN (Du and others 2007). In gene expression profiling studies of wild-type or p65/p50 double knockout (DKO) primary mouse embryonic fibroblast (MEF) cells, a subgroup of IFN-induced genes, which include GBP1 and MX1, was found to be differentially regulated by the p50 and p65 subunits of NFκB (Wei and others 2006). The IFN-induced expression of GBP1 and MX1 was blocked by treatment with IKK inhibitors. However, the subgroup of ISGs differentially regulated by IKK inhibitors in glioma cells differs from the NFκB-regulated subgroup of ISGs expressed in wild-type (WT) and p65/p50 DKO MEF cells. For example, while in p65/p50 DKO cells IFN-induced antiviral activity is enhanced, IFN-induced antiviral activity is suppressed by IKK inhibitors. One potential reason for this discrepancy may reflect cell type differences. The p65/p50 DKO MEF cells are nontransformed primary MEF cells, but the glioma cells are brain cancer cells. It is well known that transformed cells require a high constitutive NFκB activity for their survival, which is not the case in those primary nontransformed cells, which have a low basal NFκB activity (Ben-Neriah and Karin 2011). Moreover, while IFN induces relatively strong NFκB activation in MEFs, only a negligible increase in NFκB activity was induced by IFN in glioma cells (data not shown). Another possible reason is that IKK is upstream of NFκB activation in the IKK/NFκB signal transduction pathway. IKK complexes might affect other unknown factors that may influence the IFN signal transduction pathway that does not involve the NFκB transcription factor.
A number of studies have shown that many cancer cells have a variety of defects within the IFN signaling pathway, which may be one of the reasons why IFN has been successful in treating certain cancers, but has shown disappointing results in other cancers (Pfeffer and others 1998). Many cancer cells also have a defective IFN-induced antiviral pathway (Stojdl and others 2000; Stojdl and others 2003; Barber 2005; Noser and others 2007; Basagoudanavar and others 2011). For these reasons, oncolytic viruses such as VSV and EMCV have shown promise as effective agents against malignant brain tumors (Wollmann and others 2007; van den Pol and others 2009; Wollmann and others 2010). Brain tumors are among the leading causes of cancer-related deaths in the United States, with glioblastoma multiforme (GBM) being one of the most aggressive and difficult subtypes to treat. Thus, the treatment of malignant glioma is a significant clinical problem for which new strategies are desperately needed. Our results clearly show that among the glioma cells examined, the sensitivity to IFN-induced cytopathic effects is highly variable. For example, GBM6 and MT330 glioma cells are more sensitive to the antiviral action of IFN against VSV compared to U87 and SJ-G2 glioma cells. Since IKK inhibitors strongly reversed IFN-induced anticytopathic and antiviral activities in the glioma cells sensitive to the antiviral action of IFN, it is tempting to speculate that IKK inhibitors, when combined with oncolytic viruses, could provide improved anticancer activity over monotherapy with oncolytic viruses that are presently under investigation. Our results also raise the issue that IFN sensitivity is clearly context specific. While U87 and SJ-G2 glioma lines were relatively resistant to the antiviral action of IFN, these glioma lines were relatively sensitive to the IFN-induced expression of the ISG subset presently examined. Thus, future studies should be directed at defining which specific ISGs are differentially regulated by IFN in glioma cells to specifically target these ISGs to affect glioma sensitivity to viral infections.
Another important issue to be considered is that IKK inhibitors have been used in treating various autoimmune and inflammatory disorders, such as rheumatoid arthritis, lupus, multiple sclerosis, and inflammatory bowel disease. Since these inhibitors will suppress not only IFN produced in response to activation of the pattern recognition pathway, but also the antiviral action of IFN; it is possible that these IKK inhibitors will potentially enhance the susceptibility of patients with immune disorders to a variety of viral infections (Strnad and Burke 2007).
In conclusion, our results strongly suggest that IKK complexes directly regulate IFN-induced signal transduction and ISG expression. The suppression of IFN-mediated antiviral activity by IKK inhibitors opens a potential new avenue in the treatment of cancers using oncolytic viruses in combination with these IKK inhibitors.
Footnotes
Acknowledgments
We thank Dr. Lawrence Blatt (InterMune) for generously providing recombinant human IFN, and Dr. Christopher Duntsch (UTHSC, Department of Neurosurgery) and Dr. C. David James (University of California, San Francisco, Department of Neurological Surgery) for generously providing MT330 and GBM6 glioma cells, respectively. This work was supported in part by grants from the National Institutes of Health CA133322 (L.M.P. and A.M.D.), CA23099; and CA21766, a grant from the Assisi Foundation of Memphis (A.M.D.); and by the Muirhead Chair Endowment (L.M.P.) of the UTHSC and by the American Lebanese Syrian Associated Charities (A.M.D.).
Author Disclosure Statement
No competing financial interests exist.