
Abstract
Acute alcohol (ethanol) exposure is linked with increased susceptibility to infection and increased mortality in trauma and burn patients. Dendritic cells (DCs) are central mediators in innate and adaptive immune responses, and they play a role in the presentation of pathogens to adaptive immune cells. We investigated the effects of acute ethanol exposure on bone marrow-derived DC (BM-DC) responses. Total bone marrow cells, obtained from 8 to 10 week old C57BL/6 male mice, were cultured in the presence of granulocyte/monocyte-colony stimulating factor and interleukin (IL)-4 for 7 days. BM-DCs were harvested and treated with increasing doses of ethanol (50, 100, and 250 mM) at the time of, or 3 h before, lipopolysaccharide (LPS). After LPS, supernatants were collected for cytokine measurement, and cells were harvested for flow cytometry. Concurrent acute ethanol exposure and LPS treatment resulted in a dose-dependent suppression of IL-6, IL-12p40, IL-23, and IL-10. In addition, ethanol exposure before LPS dysregulated the IL-12p40/IL-23 balance and more profoundly suppressed IL-6 and IL-10 secretion by BM-DCs, as compared with cells concurrently treated with ethanol and LPS. Ethanol treatment did not affect either toll-like receptor (TLR)4 or TLR2 expression. In summary, our study demonstrates that acute ethanol exposure suppresses BM-DC LPS-induced responses, irrespective of affecting TLR4 or TLR2 expression.
Introduction
N
DCs comprise a migratory group of bone marrow-derived leukocytes that are able to uptake, transport, process, and present antigens (Hart 1997; Banchereau and Steinman 1998; Steinman 2007). After the uptake of antigen, DCs migrate from the periphery to the draining lymph nodes and undergo maturation, marked by an increased expression of major histocompatibility complex (MHC) class II and other genes that promote antigen presentation. In secondary lymphoid organs, DCs present antigen via MHC II to naïve T lymphocytes to generate T-helper (Th)1, Th2, or Th17 responses (Mazzoni and Segal 2004). Thus, in their role as APCs, DCs are a critical link between the innate and adaptive arms of the immune system.
Similar to monocytes/macrophages, DCs express various pattern recognition receptors, including toll-like receptors (TLRs). Receptor ligation of TLRs by specific ligands induces various signaling pathways to modulate immune responses. Specifically, LPS activates immune cells through its interaction with TLR4 (Takeda and others 2003), which results in the activation of multiple downstream signaling pathways and transcription factors, including the MyD88-dependent and mitogen activated protein kinase (MAPK) pathways (Takeda and others 2003). Ultimately, this leads to the release of multiple cytokines, including TNF-α, IL-1, IL-6, IL-10, IL-12, and, most recently, IL-23, which is known to be a critical mediator of Th17 polarization (Sweet and Hume 1996; Lu and others 1999; Dumitru and others 2000; Guha and others 2001; Guha and Mackman 2001; Goral and others 2004; Goral and Kovacs 2005; Liu and others 2009).
Currently, the literature addressing the effects of ethanol exposure on DC immune responses is quite limited, particularly in the context of acute ethanol exposure. To date, the effects of acute ethanol exposure on DC function are limited to the DCs derived from monocytes isolated from healthy human donors exposed to ethanol before venipuncture (Mandrekar and others 2004; Szabo and others 2004). These studies demonstrate that DCs derived from volunteers exposed to ethanol had decreased expression of costimulatory molecules CD80 and CD86, which are required for T cell activation (Mandrekar and others 2004). Functionally, the DCs from ethanol exposed volunteers induced T cell anergy, inhibited Th1 activation, and resulted in decreased T cell proliferation (Mandrekar and others 2004; Szabo and others 2004). In line with published literature on monocytes and macrophages, these studies showed decreased secretion of Th1 polarizing IL-12 and increased secretion of anti-inflammatory IL-10 (Mandrekar and others 2004). Similarly, Lau and others (2006) demonstrated that acute ethanol exposure inhibited the differentiation of bone marrow-derived DCs (BM-DCs), subsequent cosignaling molecule expression, naïve T cell activation, and impaired production of IL-12p70. Murine models of chronic ethanol abuse demonstrated that chronic ethanol consumption suppressed the ability of DCs to stimulate the activation and proliferation of naïve T cells, both in vivo and in vitro (Lau and others 2006; Eken and others 2011; Fan and others 2011). With regard to cytokine production, the DCs from ethanol-fed mice produced less IL-12p40, TNF-α, and IFN-α (Fan and others 2011). These data provide evidence which suggests that ethanol exposure results in aberrant DC immune responses, which may result in an imbalanced immune response and detrimental outcomes after infection or trauma. However, the effects of acute ethanol exposure on differentiated DCs remain to be elucidated.
To explore the impact of acute ethanol on DC cytokine production, our study utilized BM-DCs to evaluate the concurrent and pretreatment, as well as the dose-dependent effects of ethanol on DC cytokines known to mediate T cell responses, IL-12p40, IL-6, IL-23, and IL-10. Using differentiated DCs and LPS, our study models a scenario in which the host is immunologically challenged while under the influence of ethanol intoxication. Thus, our study directly measures the effects of acute ethanol exposure on differentiated DCs, rather than exploring the effects of ethanol on DC differentiation. We also sought to determine whether TLR expression contributes to ethanol-mediated cytokine dysregulation. Herein, we show that acute ethanol exposure results in a dose-dependent perturbation of IL-6, IL-12p40, IL-23, and IL-10. Moreover, our data suggest that acute ethanol exposure has more profound effects if DCs are pretreated with ethanol than if they are exposed to ethanol at the time of LPS stimulation. Lastly, we demonstrate that acute ethanol exposure blunts LPS-induced cytokine responses independent of TLR4 or TLR2 expression.
Materials and Methods
Animals
Male C57BL/6 mice (Harlan Laboratories), 8–9 weeks old, weighing 22–25 g, were used in the experiments. The animals were allowed to acclimate to the animal facility for 2 weeks before being used for experiments. All experiments were conducted in accordance with the guidelines set forth in the Animal Welfare Act and were approved by the Institutional Animal Care and Use Committee at the Loyola University Chicago Health Sciences Division.
Bone marrow isolation and DC differentiation
BM-DCs were generated in vitro from total bone marrow, as previously described (Mayordomo and others 1995; Celluzzi and others 1996; Khayrullina and others 2008). Briefly, femurs and tibias were dissected from unmanipulated mice. Both ends of each bone were cut open, and the bone marrow was flushed out with 3 mL cold RPMI-1640 medium (Thermo Fisher Scientific) using a 22-gauge needle. Subsequently, the bone marrow was brought to a single-cell suspension by pipetting and passage through a 70 μm cell strainer. Total bone marrow cells were counted by a hemocytometer, washed in RPMI medium, and adjusted to a concentration of 2×106 cells/mL. Bone marrow cells, 10×106 cells, were cultured in complete RPMI-1640 supplemented with 2 mM
Ethanol exposure and LPS stimulation in vitro
After differentiation, BM-DC (2×105 cells/well) were cultured in the presence of increasing doses of ethanol (0, 50, 100 and 250 mM) for either 3 h before, or in parallel with, LPS (1 μg/mL) stimulation in 96-well plates at 37°C and 5% CO2 for 24 h in a humidified chamber. After ethanol exposure and LPS stimulation, cell supernatants were harvested for the measurement of IL-6, IL-10, IL-12p40, and IL-23 by ELISA. In addition, the cells were harvested for measurement of cell viability (Supplementary Fig. S1; Supplementary Data are available online at
CD11c, MHC II, TLR2, and TLR4 expression
Flow cytometric analysis was performed to determine DC purity as well as TLR2 and TLR4 expression after ethanol exposure and LPS stimulation. Briefly, BM-DC (4×105 cells) were collected and resuspended in staining buffer (PBS containing 5% FCS and 2 mM EDTA). Cell suspensions were blocked with anti-CD16/32 (0.5 μg/100 μL staining buffer) and rat IgG for 20 min at 4°C and subsequently stained with PE-conjugated anti-CD11c (0.25 μg/100 μL staining buffer), FITC conjugated anti-MHC II (0.25 μg/100 μL staining buffer), PE-Cy7 conjugated anti-TLR4 (0.5 μg/100 μL staining buffer), and Alexa 647 conjugated anti-TLR2 (0.1 μg/100 μL staining buffer) for 30 min at 4°C. The cells were washed twice, resuspended in 500 μL staining buffer, and analyzed at the Loyola University Medical Center FACS Core Facility using an FACSCantoI (BD Bioscience). All FACS data analyses were performed using FlowJo software (Treestar). All antibodies used for flow cytometry were purchased from eBioscience.
Cytokine measurement
Cytokine production in BM-DC cultures was determined by ELISA. The IL-6 and IL-12p40 ELISAs were purchased from BD Bioscience, and the IL-10 and IL-23 ELISAs were purchased from R&D Systems. Both IL-12 and IL-23 are members of the IL-12 family that share the p40 subunit (Murphy and others 2003; Langrish and others 2005). Given that we measured IL-23 by an ELISA kit that does not react with IL-12, we used IL-12p40 as a proxy for IL-12.
Statistical analysis
Results are presented as mean±standard error of the mean. ANOVA with Tukey post hoc testing was used to compare the experimental groups. All statistical analyses were performed using InStat 3.0 (GraphPad Software). P<0.05 was considered statistically significant.
Results
DC purity
DCs were differentiated from total bone marrow cells in the presence of GM-CSF and IL-4, as previously described (Mayordomo and others 1995; Celluzzi and others 1996). After differentiation, BM-DC purity was determined based on the coexpression of DC markers CD11cand MHC II (Liu 2001; Shortman and Liu 2002; Khayrullina and others 2008). The culture of total bone marrow cells with GM-CSF and IL-4 yielded an average of >90%CD11c+MHC II+ cells (Fig. 1).
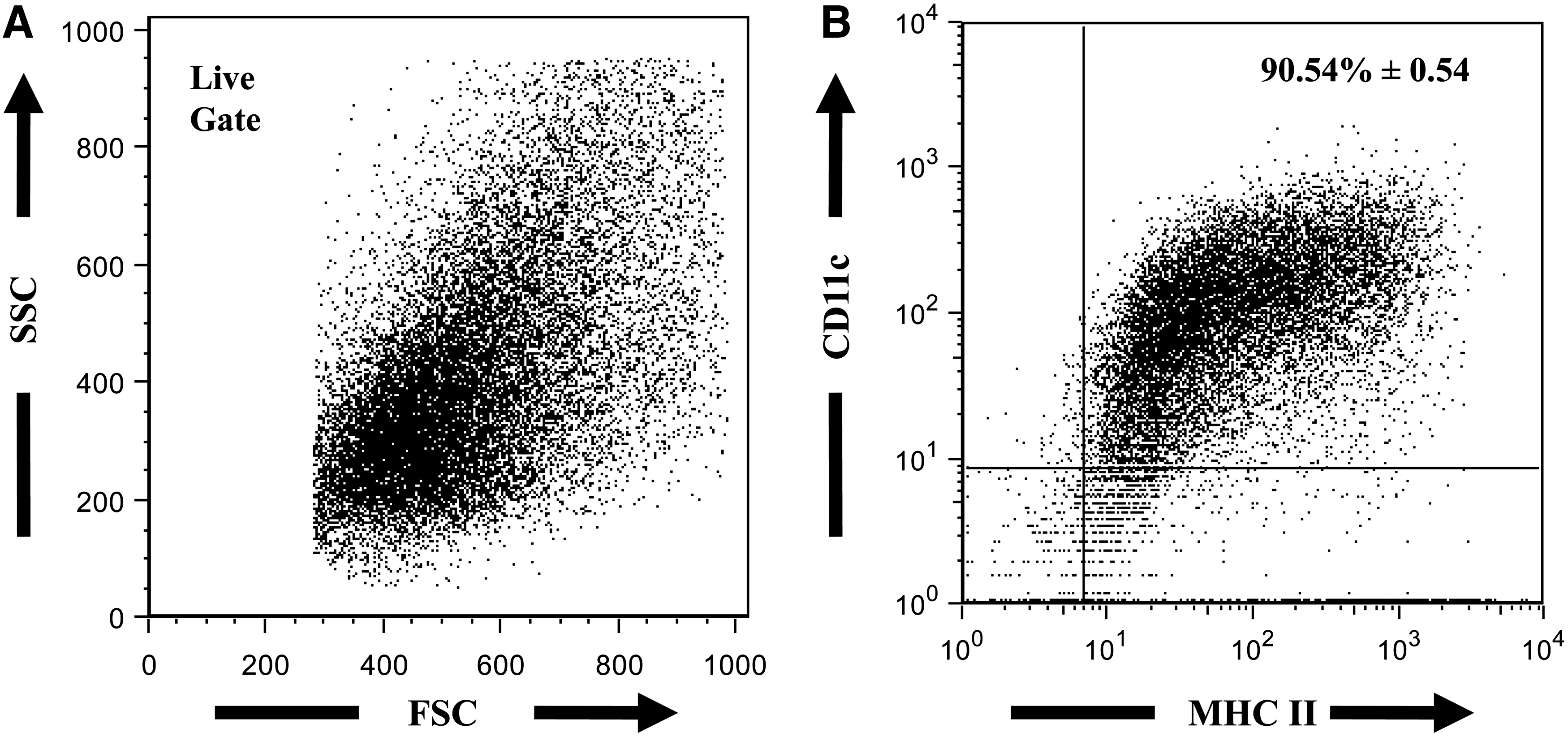
Representative FACS plots of BM-DC population. After 7–10 day culture in complete RPMI media containing granulocyte/monocyte-colony stimulating factor (50 ng/mL) and IL-4 (100 U/mL), cells were harvested and analyzed for the expression of DC markers, CD11c and MHC II. Cell suspensions were stained with FITC-labeled anti-MHC II and PE-labeled CD11c antibodies.
Ethanol exposure leads to BM-DC cytokine dysregulation
To determine the effect of ethanol exposure on the secretion of pro- and anti-inflammatory cytokines, BM-DCs were cultured with LPS (1 μg/mL) in the presence of increasing doses of ethanol (50, 100 and 250 mM) or after 3 h of ethanol pretreatment at the same doses (Fig. 2). We evaluated IL-12p40, IL-6, and IL-23, 3 pro-inflammatory cytokine generated by DCs that are important for Th1 (IL-12) and Th17 (IL-6 and IL-23) polarization. We also examined IL-10, an anti-inflammatory cytokine associated with a Th2 immune response. Cell viability by flow cytometry, as determined by the percent of cells in the live gate, was not affected by ethanol or LPS treatment (Supplementary Fig. S1).

BM-DC cytokine release after ethanol exposure and LPS stimulation. BM-DCs (2×106 cell/well) were cultured in 96-well plates in the presence of LPS (1 μg/mL) and varying concentrations of ethanol for 24 h at 37°C. Supernatants were harvested for the measurement of IL-6
The concurrent treatment of BM-DC with LPS and ethanol led to a dose-dependent decrease in IL-12p40, IL-23, and IL-10 with a maximum suppression of IL-6 seen at 50 mM (Fig. 2). The introduction of LPS and 50 mM ethanol in parallel resulted in a 25% decrease in IL-6 secretion from BM-DCs compared with LPS alone (Fig. 2A, P<0.05 for 50 mM ethanol+LPS group as compared with LPS alone). At increasing doses, no additional suppression was seen, though it should be noted that there was no statistical significance between the 3 ethanol doses with regard to IL-6 secretion. This ethanol-mediated cytokine suppression was also seen with IL-23 (Fig. 2C, P<0.001 as compared with LPS alone). Specifically, the concurrent addition of LPS and ethanol (50, 100, and 250 mM) resulted in ∼40%, 55%, and 68% reduction of IL-23 release, respectively. In response to concurrent ethanol exposure and LPS stimulation, IL-12p40 secretion was decreased as compared with the LPS alone group, but only reached significance when treated with 250 mM ethanol (Fig. 2B, P<0.05). While 50 mM ethanol treatment administered simultaneously with LPS did not affect IL-10 secretion, 100 and 250 mM ethanol exposure plus LPS stimulation resulted in 30% and 75% reductions in IL-10 release, respectively (Fig. 2D, P<0.05 compared with LPS alone). Co-culture with 250 mM ethanol and LPS resulted in a significant decrease in IL-10 release as compared with co-cultures with 50 and 100 mM ethanol (Fig. 2D, P<0.05). Together, these data suggest that ethanol treatment at the time of immune challenges leads to global BM-DC cytokine suppression.
To further elucidate whether ethanol exerts differential effects on BM-DC cytokine secretion after immune stimulation, BM-DCs were pretreated with ethanol for 3 h before culture with LPS. Pretreatment of BM-DCs with increasing doses of ethanol (50, 100, and 250 mM) resulted in ∼25%, 40%, and 60% decreased IL-6 secretion in response to LPS, respectively (Fig. 2A, P<0.05, as compared with LPS alone group). Moreover, the pretreatment of DCs with 250 mM ethanol resulted in a 50% reduction in IL-6 release as compared with BM-DCs pretreated with ethanol 50 mM (P<0.05) and with DCs concurrently cultured with LPS and ethanol 250 mM (P<0.05). Ethanol treatment before LPS stimulation leads to a marked dose-dependent suppression (∼30%, 45%, and 75% at 50, 100, and 250 mM ethanol, respectively) of IL-23 as compared with LPS alone (Fig. 2C, P<0.001). However, this suppression was irrespective of the timing of ethanol exposure relative to LPS treatment and was most profound in the cells exposed to 250 mM ethanol as compared with lower doses (Fig. 2C, P<0.05). While these 2 cytokines exhibited a similar, or slightly more extensive, immune suppression with ethanol pretreatment, IL-12p40 was paradoxically elevated in supernatants from BM-DCs treated with 50 mM ethanol before LPS stimulation, as compared with LPS treatment alone (Fig. 2B, P<0.05). Likewise, compared with BM-DCs treated with 50 and 100 mM ethanol in parallel with LPS, IL-12p40 secretion from corresponding pretreated cells was significantly elevated (P<0.05). However, the pretreatment of cells with higher ethanol concentrations (100 and 250 mM) did not lead to significant changes in IL-12p40 production when compared with LPS treatment alone. These data suggest the ethanol may exert differential effects on the mechanisms that modulate these cytokines, and may alter the ability of DCs to effectively induce one T cell subset over another. The pretreatment with ethanol also seems to more profoundly impact the ability of BM-DC to exert immunosuppressive effects. Prior exposure of BM-DCs with 50, 100, and 250 mM ethanol resulted in a 60%, 81%, and 100% decline in IL-10 release, respectively (Fig. 2D, P<0.05 as compared with LPS alone). When compared with BM-DCs exposed to LPS and ethanol concurrently, ethanol exposure 3 h before LPS exacerbated the suppression of IL-10 secretion. Prior incubation with 50 and 100 mM, ethanol diminished IL-10 release by 56% and 73%, as compared with BM-DCs at the respective doses in the concurrent treatment group (Fig. 2D, P<0.05). Notably, the pretreatment of DCs with 250 mM ethanol abolished IL-10 release, whereas the coculture with ethanol and LPS diminished IL-10 release by 75% (Fig. 2D, P<0.05, as compared with 250 mM EtOH plus LPS group).
Ethanol does not alter BM-DC TLR4 or TLR2 expression
In order to determine whether the difference in cytokine levels with ethanol exposure were a result of alterations in the receptor for LPS, we used flow cytometry to analyze the expression of TLR4 on BM-DCs (Fig. 3). LPS stimulation resulted in a ∼25% decrease in the percentage of DCs expressing TLR4, as compared with unstimulated cells (Fig. 3A, P<0.05). Similarly, LPS challenge resulted in a ∼50% decrease (P<0.05) in the mean fluorescence intensity (MFI) of TLR4, as compared with unstimulated cells (Table 1). This reduction in percentage as well as per cell expression of TLR4 expression (summarized in Table 1) may be due to receptor ligation or internalization. DCs exposed to ethanol, either before or at the time of LPS challenge, demonstrated a similar reduction in the percentage and per cell expression of TLR4 (Fig. 3), suggesting that ethanol does not influence the levels of TLR4 after LPS stimulation. Together, these data suggest that the cytokine changes found after ethanol exposure are independent of TLR4 expression and may suggest aberrant downstream signaling.

TLR4 expression on DCs. BM-DCs (2×106 cell/well) were cultured in 96-well plates in the presence of LPS (1 μg/mL) and varying concentrations of ethanol for 24 h at 37°C. Cells were harvested, and cell suspensions were stained with PE-conjugated anti-CD11c, FITC-conjugated anti-MHC II, and PE-Cy7 conjugated anti-TLR4. Representative FACS plots were taken from a single animal (ethanol dose–50 mM), and demonstrate the percentage of TLR4+ cells
MFIs are expressed as fold difference in order to combine data obtained from independent experiments. Data are represented as means±SEM, n=4–7 animals/group from 2 independent experiments.
P<0.05 as compared with unstimulated, ethanol 50, and 100 mM groups by ANOVA with Tukey post hoc test.
P<0.05 as compared with unstimulated and ethanol-only groups by ANOVA with Tukey post hoc test.
P<0.0001 as compared with unstimulated and ethanol-only groups by ANOVA with Tukey post hoc test.
P<0.05 as compared with ethanol 250 group by ANOVA with Tukey post hoc test.
LPS, lipopolysaccharide; TLR, toll-like receptor; MHC, major histocompatibility complex; SEM, standard error of the mean.
LPS has been reported to induce the expression of TLR2 via the activation of TLR4-dependent pathways (Faure and others 2001; Fan and others 2003; Wu and others 2011). To assess whether ethanol exposure affects LPS induction of TLR2 expression, we determined the effects of ethanol on BM-DC expression of TLR2 by flow cytometry (Fig. 4). Though LPS stimulation alone significantly increased the percentage of DCs expressing TLR2 by 2.3-fold as compared with unstimulated cells (Fig. 4A, P<0.05), ethanol-treated BM-DCs also demonstrated similar increases in the percent of TLR2+ DCs. Similarly, LPS challenge heightened the MFI of TLR2, as compared with unstimulated cells (Table 1, P<0.0001). Neither paradigm of ethanol exposure altered the MFI of TLR2, as compared with BM-DCs cultured with LPS alone. These results are summarized in Table 1 and indicate that ethanol exposure does not perturb LPS-induced TLR2 expression.

TLR2 expression on DCs. BM-DCs (2×106 cell/well) were cultured in 96-well plates in the presence of LPS (1 μg/mL) and varying concentrations of ethanol for 22 h at 37°C. Cells were harvested, and cell suspensions were stained with PE-conjugated anti-CD11c, FITC-conjugated anti-MHC II, and Alexa-647-conjugated anti-TLR2. Representative FACS plots were taken from a single animal (ethanol dose–50 mM), and demonstrate the percentage of TLR2+ cells
Discussion
The results presented in this article clearly demonstrate that exposure to ethanol suppresses BM-DC secretion of pro- and anti-inflammatory cytokines which contribute to T-cell polarization, in a manner independent of TLR4 and TLR2 expression. These effects are dose dependent, and are further influenced by the timing of alcohol administration relative to the LPS challenge. Despite the differential cytokine profiles depending on the treatment paradigm used, neither ethanol pretreatment nor concurrent culture with ethanol and LPS altered TLR4 and TLR2 expression. While we recognize that our studies were carried out in vitro and may have limited physiological implications, our results provide strong evidence which suggests that ethanol exposure suppresses BM-DC immune response in a dose-dependent manner.
The activation of cytokine production and secretion in response to LPS, the main component of the outer membrane of Gram-negative bacteria, predominantly depends on TLR4 signaling. When combined with the LPS-binding protein, LPS is transferred to TLR4 by CD14, making CD14 a critical adaptor molecule for TLR signaling. In addition, LPS ligation to TLR4 requires accessory protein MD-2 to activate TLR4 signaling and induce the transcription of immune response genes (Guha and Mackman 2001; Nagai and others 2002; Lorne and others 2010).
Since LPS predominately affects changes in cytokine production via the TLR4 pathway, and indirectly via TLR2, we hypothesized that the cytokine dysregulation observed in our study may by correlated with altered expression of these pattern recognition receptors. Earlier, several groups demonstrated that in vivo and in vitro acute ethanol exposure disrupts macrophage TLR4/CD14 clustering by interfering with lipid rafts and actin cytoskeleton reorganization after LPS stimulation (Dai and others 2005; Dai and Pruett 2006a, 2006b). Our study directly examined the effect of ethanol on TLR expression. Though no alteration in TLR expression was observed in our study, it is possible that these mechanisms play a role in the cytokine changes found in DCs after ethanol exposure and LPS challenge. Additional experiments will explore TLR4/CD14 clustering and cytoskeleton reorganization in response to LPS to answer whether ethanol exerts its immunosuppressive effects on DCs by inhibiting proper receptor interactions and/or subsequent downstream signaling. Moreover, our study found that LPS-induced expression of TLR2 was not affected by the presence of ethanol; yet, we did not directly test the functionality of TLR2. Future experimentation with TLR2-specific agonists will elucidate whether ethanol abrogates TLR2-dependent immune responses.
In our study, we also sought to determine how the timing of ethanol exposure relative to LPS stimulation affects cytokine secretion. For the Th1 polarizing cytokine IL-12, ethanol exposure (50 mM) before LPS stimulation generated significantly higher IL-12 levels compared with LPS alone and those cells treated concurrently. This is an interesting observation, suggesting how ethanol interacts with cellular machinery to regulate cytokine production may be time dependent. Of note, Mandrekar and others (2004) and Lau and others (2006) demonstrated that ethanol exposure perturbs the differentiation of DCs and subsequent IL-12 production. In contrast to our study, these studies used ethanol during the differentiation process, potentially altering additional cell subsets that may impact the ability of DC precursors to effectively differentiate into functional DCs. Moreover, in vivo exposure to a single dose of ethanol (2.9 g/kg) results in decreased LPS-induced IL-12 production from splenic macrophages, 6 hrs after an ethanol injection (Karavitis and others 2008). Taken together, these results suggest that the stage of immune cell differentiation and timing of ethanol exposure may be important factors in driving ethanol-induced perturbation of IL-12p40 secretion. Further, these data suggest that ethanol exposure may skew the ability of DCs to aid in T cell polarization after immune challenge.
With reference to IL-6 and IL-23, DC secreted cytokines that work in concert to drive naïve T cells toward the Th17 phenotype, the pretreatment with ethanol exerted a more robust cytokine suppression after LPS stimulation as compared with co-cultured cells. As previously mentioned, IL-23 is a member of the IL-12 family, and both cytokines share the p40 subunit. The role of the p40 subunit has long been described as being crucial for the biological functions of IL-12, as the IL-12R recognizes the heterodimeric structure. However, until the discovery of the IL-23 p19 subunit, it was thought that excess p40 formed homodimers, which exert antagonistic effects on IL-12 (Gately and others 1998). It is now understood that the p40 subunit may combine with p35 to form IL-12p70, p19 to form IL-23 or form homodimers (Gately and others 1998; Oppmann and others 2000; Khader and others 2006). Today, IL-23 is recognized for its role in promoting the production of Th17 effector cytokines IL-17 and IL-22 (Aggarwal and others 2003; Langrish and others 2005; Liang and others 2006). Interestingly, in BM-DCs treated with ethanol before LPS culture, there was a dramatic decrease in IL-23 with a relative increase in IL-12. On the other hand, concomitant treatment with ethanol and LPS resulted in a reduction of both cytokines. These data may suggest that ethanol exposure in advance of an immune challenge may preferentially shift the utility of the p40 subunit toward IL-12 production. Although not examined in this study, others have shown that LPS/TLR4-mediated induction of IL-23 p19 expression depends on NF-κβ and AP-1 (Liu and others 2009), which are modulated by MyD88 and MAPK pathways, respectively. With regard to AP-1, Liu and others (2009) were the first who established that AP-1acts to promote the expression of IL-23 p19 under the direction of ERK. Of note, ERK is also a known regulator of the p40 subunit of IL-23, the subunit that is shared with IL-12; however, while ERK promotes the expression of p19, it inhibits the expression of p40 (Feng and others 1999), suggesting that ethanol may impact ERK activity and the relative expression of these subunits.
Alterations in ERK activity, in conjugation with other members of either the MAPK or MyD88 pathway, have been attributed to alterations in IL-6 after acute ethanol treatment. In vivo acute ethanol exposure reduces LPS-induced IL-6 production from splenic macrophages (Goral and others 2004; Goral and Kovacs 2005; Karavitis and others 2008). Goral and others further demonstrated that ethanol modulates IL-6 by transiently down-regulating the activation of p38 and ERK in unstimulated and LPS challenged splenic macrophages (Goral and others 2004; Goral and Kovacs 2005). Our results indicate that when pretreated with ethanol, LPS-induced DC IL-6 production is suppressed in a dose-dependent manner, suggesting that ethanol modulates IL-6 release in fully differentiated DCs. While further experimentation is needed to determine the effect of ethanol on p38 and ERK pathways in BM-DCs in response to LPS, these previous studies support the idea that ethanol decreases ERK activity. As just mentioned, this ethanol-mediated reduction in ERK activity could explain the diminished secretion of the Th17 polarizing cytokines IL-6 and IL-23, and the elevation of Th1 skewing IL-12p40 in a time-dependent manner. The temporal role of ERK in DC cytokine production as well as other functional measures of DC activity will be the focus of future studies.
While our pro-inflammatory cytokine data support previous findings regarding ethanol and immune cell subsets, it is clear that more research needs to be conducted to examine the impact of ethanol on DC immune function. Moreover, even less is understood about the effects of ethanol on DC anti-inflammatory profiles. Current literature focusing on DC IL-10 production is limited to studies of human peripheral blood monocyte-derived DCs. In their study, Mandrekar and others (2004) report an increase in LPS-induced IL-10 from DCs generated in the presence of ethanol. While the range of IL-10 levels we obtained correlate with these data, we demonstrate a dose-dependent suppression of LPS-induced IL-10, which is further exacerbated if DCs are pretreated with ethanol before LPS stimulation. Moreover, Mandrekar and others (2004) demonstrated increased IL-10 from DCs generated in the presence of ethanol, irrespective of LPS stimulation. In our studies, we found that ethanol alone does not induce a detectable level of IL-10. These disparities in our data may be due to differences in cell population and differentiation methods as previously discussed. Together, these data imply that ethanol plays a differential role in anti-inflammatory cytokine production, which may be due to the impact of ethanol on DC differentiation and maturation.
In conclusion, our study demonstrates that acute ethanol exposure dysregulates LPS-induced immune responses irrespective of affecting TLR4 or TLR2 expression. These effects also appear to be related to the timing of ethanol exposure relative to immune challenge, as our data suggest that ethanol exposure before LPS impairs IL-23 and IL-6 secretion, and shunts the immune response toward an IL-12 response. While further studies need to be conducted to elucidate the immunomodulatory mechanisms altered by ethanol in DCs, the ethanol-induced cytokine dysregulation observed in this study is an important consideration when investigating the role of ethanol in DC/Th17 cell-mediated pathologies such as rheumatoid arthritis and inflammatory bowel disease.
Footnotes
Acknowledgments
This study is supported by NIH grants R01AA015731 (MAC) and R01AA015731-04S1 (MAC). Juan L. Rendon is supported by NIH grants F30AA020167 (JLR), T32AA013527 (EJK), and the Loyola University Chicago Stritch School of Medicine Combined MD/PhD Program.
Author Disclosure Statement
All authors declare that no competing financial interests exist.