
Abstract
Cytokine inducible SH2-containing protein (CISH), which negatively regulates cytokine signaling by inhibiting JAK2/STAT5 activity, is regarded as a therapeutic target for inflammatory diseases. Farnesoid X receptor (FXR), a ligand-activated transcription factor, has been proposed to play a protective function in the inflammatory responses. However, the role of FXR in modulation of CISH expression is unknown. In the present study, we for the first time identified that in human hepatoma cell line HepG2 the activation of FXR by the natural agonist chenodeoxycholic acid (CDCA) and the synthetic specific agonist GW4064 upregulated CISH at both transcriptional and translational levels, and inhibited interleukin (IL)6-induced STAT5 activation. Moreover, the in vivo experiment demonstrated that gavaging mice with CDCA increased CISH expression and reduced basal STAT5 phosphorylation in liver tissues. Reporter assay showed that FXR agonists enhanced the transcriptional activity of CISH promoter. These data suggest that FXR may serve as a novel molecular target for manipulating CISH expression in hepatocytes. FXR-mediated upregulation of CISH may play an important role in the homeostasis of cytokine signal networks and be beneficial to control cytokine-associated inflammatory diseases.
Introduction
C
To prevent detrimental effects, the intensity and duration of JAK–STAT activation are tightly regulated. Tyrosine phosphatases, receptor internalization, and members of the protein inhibitors of activated STAT family contribute to the negative regulatory network. Recently, the discovery of the protein family of suppressors of cytokine signaling (SOCS) has highlighted an important central mechanism (Starr and Hilton 1998). SOCS that can negatively regulate the response of immune cells either by inhibiting JAK/STAT activity or by competing with signaling molecules for binding to the phosphorylated receptor (Kile and Alexander 2001; Krebs and Hilton 2001) are now considered as important regulators of normal physiology and complicated cytokine signal networks in various kinds of diseases (Diamond and others 2000; Tan and Rabkin 2005; Ramgolam and Markovic-Plese 2011). Interestingly, some studies suggested that the anti-inflammatory effects of some nuclear receptors such as proliferator-activated receptors (PPAR) β protecting the diabetic kidney against ischemia/reperfusion injury and PPAR γ attenuating brain inflammation were associated with the induction of SOCS and the inhibition of JAK–STAT activity (Collino and others 2011; Park and others 2003).
Another member of nuclear receptor superfamily, farnesoid X receptor (FXR, NR1H4) has also been proposed as a novel molecular target in inflammatory diseases (Wang and others 2009; Gadaleta and others 2011). FXR, which is highly expressed in liver, intestine, and kidney (Forman and others 1995) can be activated by several bile acids (BAs) such as chenodeoxycholic acid (CDCA) (Makishima and others 1999) and a wide variety of compounds like androsterone, fexaramine, and synthetic agonist GW4064 (Maloney and others 2000). Although FXR was originally shown to play a key role in the maintenance of cholesterol and BA homeostasis, some ligands of FXR have recently been reported to exert anti-inflammatory actions and decrease the inflammation gene expression by several mechanisms, including the formation of direct complexes with activator protein-1 and nuclear factor-κB family members and/or the modulation of mitogen-activated protein kinase activity (He and others 2006; Wang and others 2008a, 2008b). But so far there is no final conclusion about whether there is a link between the anti-inflammatory action of FXR agonists and the induction of SOCS.
Cytokine inducible SH2-containing protein (CISH or CIS) was the first member of the SOCS family to be described (Yoshimura and others 1995). CISH is present in cells at very low levels but can be generally rapidly induced by a set of cytokines that activate STAT5, such as IL2, IL3, IL6, EPO, and growth hormone (Jegalian and Wu 2002). Then the increased expression of CISH blocks the cytoplasmic docking and activation of STAT5 and thereby inhibits downstream cytokine signaling (Matsumoto and others 1997, Starr and others 1997; Hanada and Yoshimura 2002; Larsen and Ropke 2002). Thus, CISH is considered as a specific inhibitor of cytokine-STAT5 pathway. However, little is known about a role of nuclear receptors in modulation of CISH expression.
In this study, we investigated that the CISH expression was significantly increased in hepatocyte HepG2 and mice liver tissues in response to FXR ligand(s), which provides an additional link between FXR and inflammatory cytokines. Additionally, the 2 agonists for FXR could markedly suppress pro-inflammatory cytokine expression and IL6-stimulated STAT5 activation. Results from our data suggest that the anti-inflammatory effect of FXR in hepatocyte may be relevant to the upregulation of CISH.
Materials and Methods
Materials
CDCA, GW4064 (2 agonists for FXR), and guggulsterone (GS, an antagonist for FXR) were purchased from Sigma Chemical Company (St. Louis, MO). Lipofectamine2000, TRIzol reagent, M-MLV reverse transcriptase, and all products for cell culture were bought from Invitrogen (Carlsbad, CA). Oligo (dT) primer, luciferase assay system, β-galactosidase assay system, and the firefly luciferase reporter vector pGL3-basic were from Promega (Madison, WI).
Cell culture
Human hepatoma cell line HepG2 was maintained in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (complete medium) at 37°C and 5% CO2. The cells grown to 60%–80% confluence in 6-well plates were treated with different concentrations of FXR agonists and/or antagonist and cultured in Opti-modification of Eagle's Minimal Essential Medium supplemented with 0.5% FBS and without antibiotics (conditioned medium).
Animal experiments
C57BL/6 mice (aged 5–7 weeks, 6 mice/group) purchased from the Laboratory Animal Center of the Third Military Medical University were housed in a temperature-controlled room and had free access to food and water. Subsequently, the mice were gavaged once a day with either vehicle alone (soyabean oil) or vehicle containing different dose CDCA (10 or 50 mg/kg) for 1 week. The mice were fasted overnight before livers removed for RNA and protein extractions.
Reverse transcription–quantitative polymerase chain reaction
Total RNA was extracted with TRIzol reagent and the first-strand cDNA was synthesized using reverse transcriptase. The reverse transcription–quantitative polymerase chain reaction (RT-qPCR) was performed using Bio-Rad iQ5 Gradient Real Time PCR system according to the manufacturer's instruction, taking β-actin as internal control. The primers specific for SHP, CISH, IL6, tumor necrosis factor α (TNFα), and β-actin were listed in Table 1.
Western blot
The HepG2 cells or mice liver tissues were washed twice with phosphate-buffered saline prior to lysis in lysis buffer containing protease inhibitor. Protein concentrations were determined by Bradford protein assay. Western blot analysis was performed and β-actin was used as corresponding loading control. Rabbit anti-CISH antibody (reactive with both human and mouse CISH) was from Abcam (San Francisco, CA), rabbit anti-phosphorylated-STAT5 (p-STAT5) and anti-total-STAT5 (t-STAT5) antibody were purchased from Cell Signaling Technology (Beverly, MA). Mouse anti-β-actin antibody (reactive with both human and mouse β-actin) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). HRP-labeled goat anti-mouse IgG and the ECL chemiluminescence kit were from Pierce (Rockford, IL).
Plasmid construction
Human CISH promoter region containing fragment (−3009∼+199) was amplified by PCR with the oligonucleotides 5′-gc
Transfection and reporter assays
HepG2 cells were plated in 48-well plates and grown at 60%–80% confluency for transfection using Lipofectamine2000 with luciferase reporter pGL3-CISH in the presence or absence of pCMX-vpFXR, and pCMX was added to ensure identical amounts of DNA in each well. Transfection efficiency was monitored by co-transfection of pCMV-β-galactosidase plasmid. Five hours after transfection, the cells were treated with FXR ligands or vehicle (0.1% dimethylsulfoxide) in conditioned medium. Following 24 h incubation, cells were harvested and the activity assays of luciferase and β-galactosidase were performed according to the manufacturer's instructions. Luciferase activity was then normalized against β-galactosidase activity. Transfection experiments were done at least 3 times in triplicate. Data were represented as fold induction over reporter gene alone.
Statistical analysis
All data were expressed as mean±standard deviation unless otherwise stated. Comparisons between 2 groups were made with unpaired Student's t-test. Non-parametric comparisons between 3 or more groups were made with analysis of variance followed by Kruskal–Wallis post hoc analysis. P<0.05 was considered statistically significant.
Results
FXR is functional in HepG2 cells
SHP is one of the FXR target genes induced after FXR activation, so its transcriptional level can reflect the function of FXR. As shown in Fig. 1A, treatment with natural ligand CDCA and specific ligand GW4064 led to increase in the mRNA levels of SHP in a dose-dependent manner, suggesting that FXR expressed in HepG2 cells is biologically active.
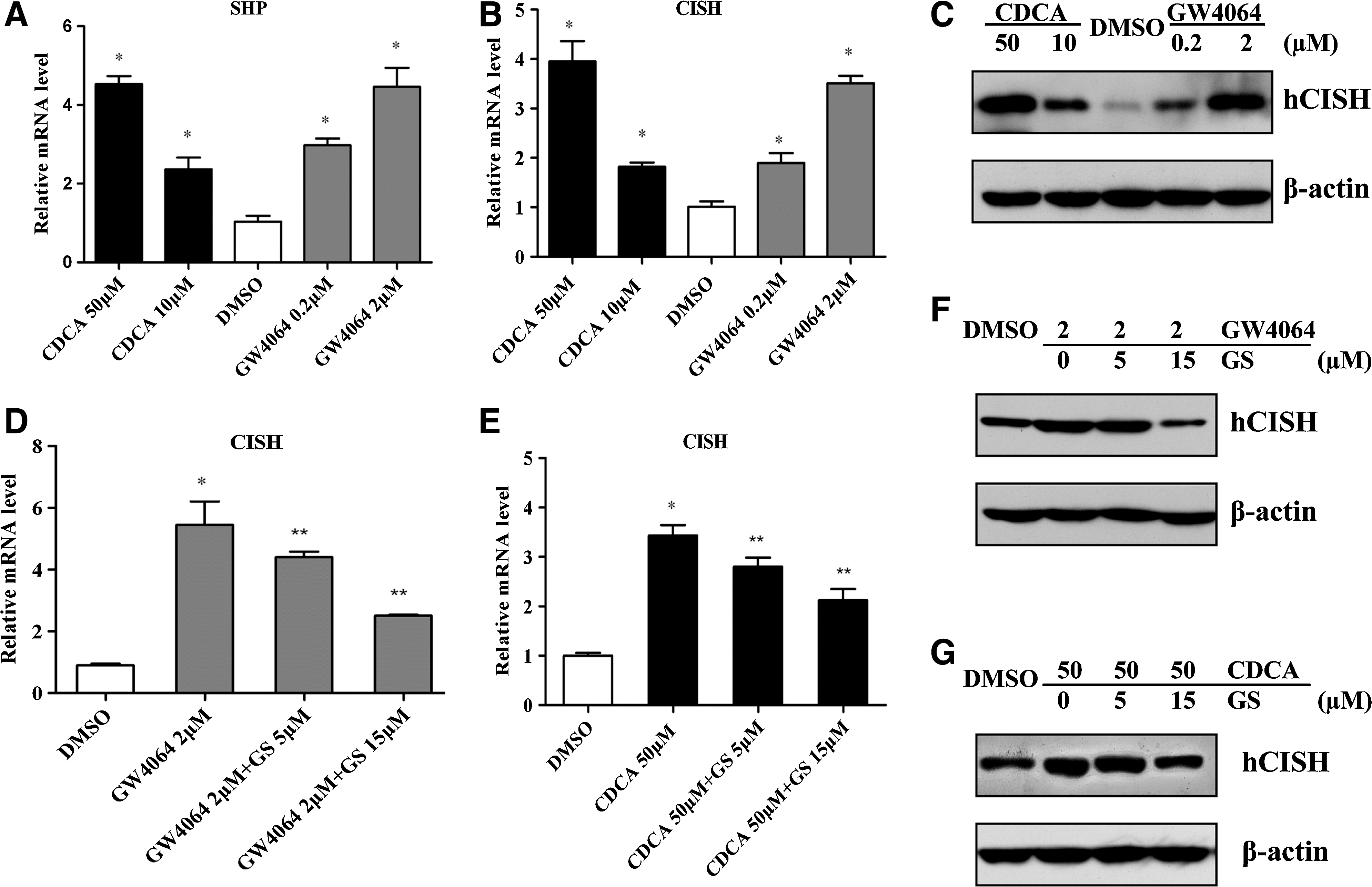
GW4064 or CDCA-mediated upregulation of CISH expression in hepatocyte. HepG2 cells were grown to about 70%–80% confluence in the complete medium and then cultured in the conditioned medium in the presence of various concentrations of FXR ligands (GW4064 or CDCA) or vehicle (0.1% DMSO) for 24 h.
Treatment with FXR agonists leads to upregulation of CISH in HepG2 cells
We then analyzed the effect of FXR ligands on CISH expression. Figure 1B showed that CDCA and GW4064 treatment could dose-dependently increase CISH mRNA level in HepG2 cells as revealed by RT-qPCR. The FXR agonists-induced CISH protein expression was also confirmed by Western blot (Fig. 1C). But pretreatment with GS (an FXR antagonist) could substantially inhibit CDCA or GW4064-mediated upregulation of CISH, which further confirmed the role of FXR in regulation of CISH (Fig. 1D–F).
CDCA induces CISH expression and inhibits STAT5 phosphorylation in vivo
To investigate the role of FXR on CISH expression in vivo, C57BL/6 mice were administered CDCA (10 or 50 mg/kg/day) for a week and the liver tissues were extracted to analysis for SHP and CISH levels. Figure 2A presented that CDCA treatment enhanced SHP expression at the transcriptional level, denoting that FXR also can be activated by CDCA in vivo. We then found FXR activation could stimulate CISH in mice liver at both mRNA level (Fig. 2B) and protein level (Fig. 2C) in a dose-dependent manner. Because CISH is considered as a specific endogenous inhibitor of STAT5 pathway, we then investigated whether FXR ligands could affect STAT5 activation. As expected, CDCA administration could reduce the phosphorylation of STAT5 (Fig. 2C), indicating that upregulation of CISH by FXR has a negative role on STAT5 signaling.
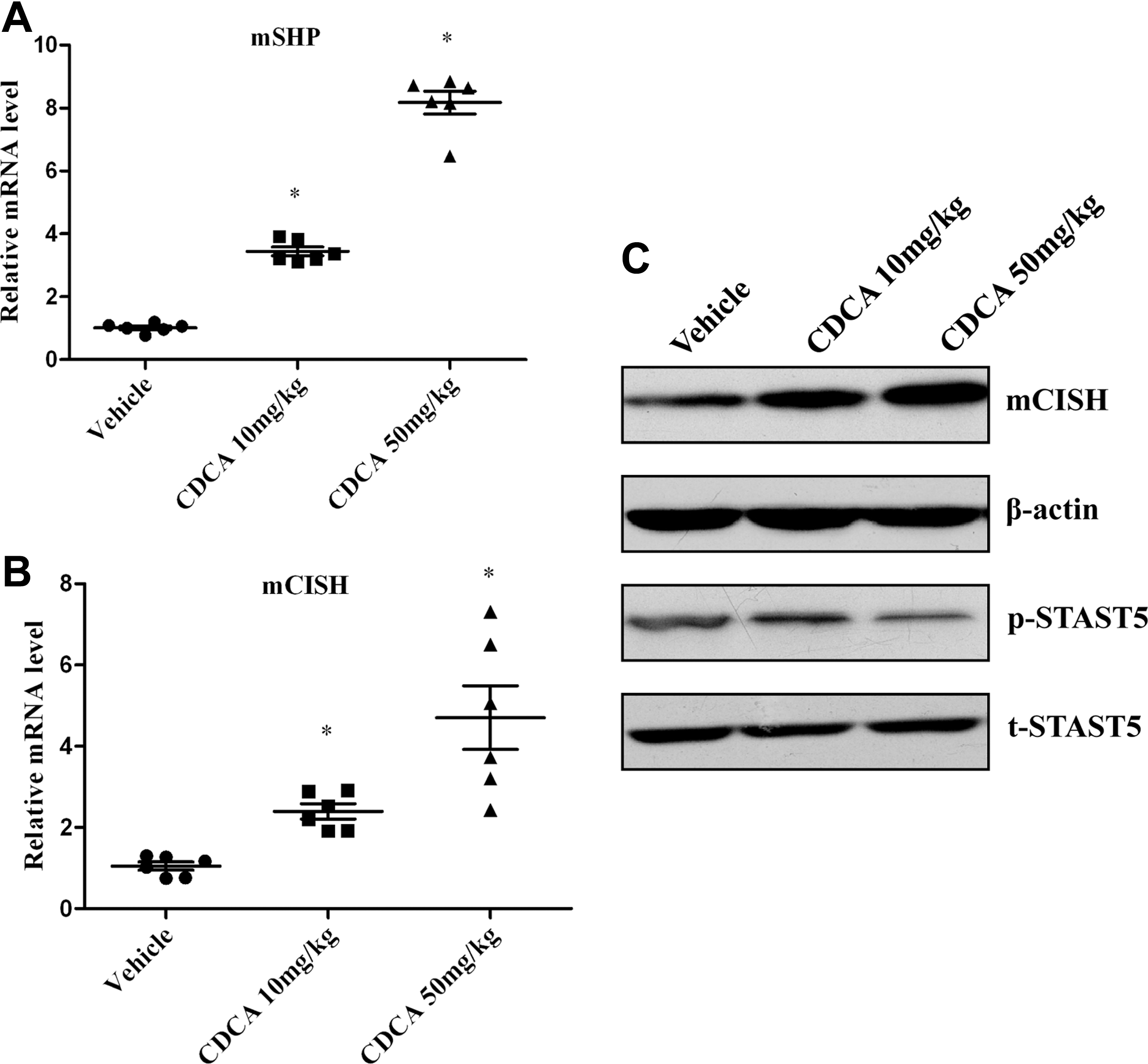
CDCA administration increases CISH expression and inhibits STAT5 activation in liver tissues of C57BL/6 mice. Mice were gavaged once a day with either vehicle alone (soyabean oil) or different dose CDCA (10 or 50 mg/kg/day) for 1 week. The mice were fasted overnight before livers removed for RNA and protein extractions.
FXR ligands repress pro-inflammatory cytokine expression and inhibit IL6-induced STAT5 activation in HepG2 cells
We then investigated the anti-inflammatory effects of FXR. By RT-qPCR analysis (Fig. 3A), a decrease in the mRNA level of pro-inflammatory cytokines such as IL6 and TNFα was detected after FXR activation in HepG2 cells, indicating that FXR plays an important role in regulation of the pro-inflammatory genes expression. We also investigated the effect of FXR in the STAT5 activation in HepG2 cells. Cells were treated with FXR ligands for 24 h and subsequently stimulated with pro-inflammatory cytokine IL6 for an additional 1 h. We then prepared the cell extracts and assayed for STAT5 phosphorylation. The Western blot results illuminated that stimulation with IL6 resulted in activation of STAT5, but it was significantly abated by pretreatment with CDCA or GW4064 (Fig. 3B, C). These effects were accompanied by an increased expression of CISH as revealed by Western blots and RT-qPCR (Fig. 3B–D). These results show that FXR agonists CDCA and GW4064 may exert inhibitory actions on cytokine-STAT5 inflammatory signaling via upregulation of CISH.
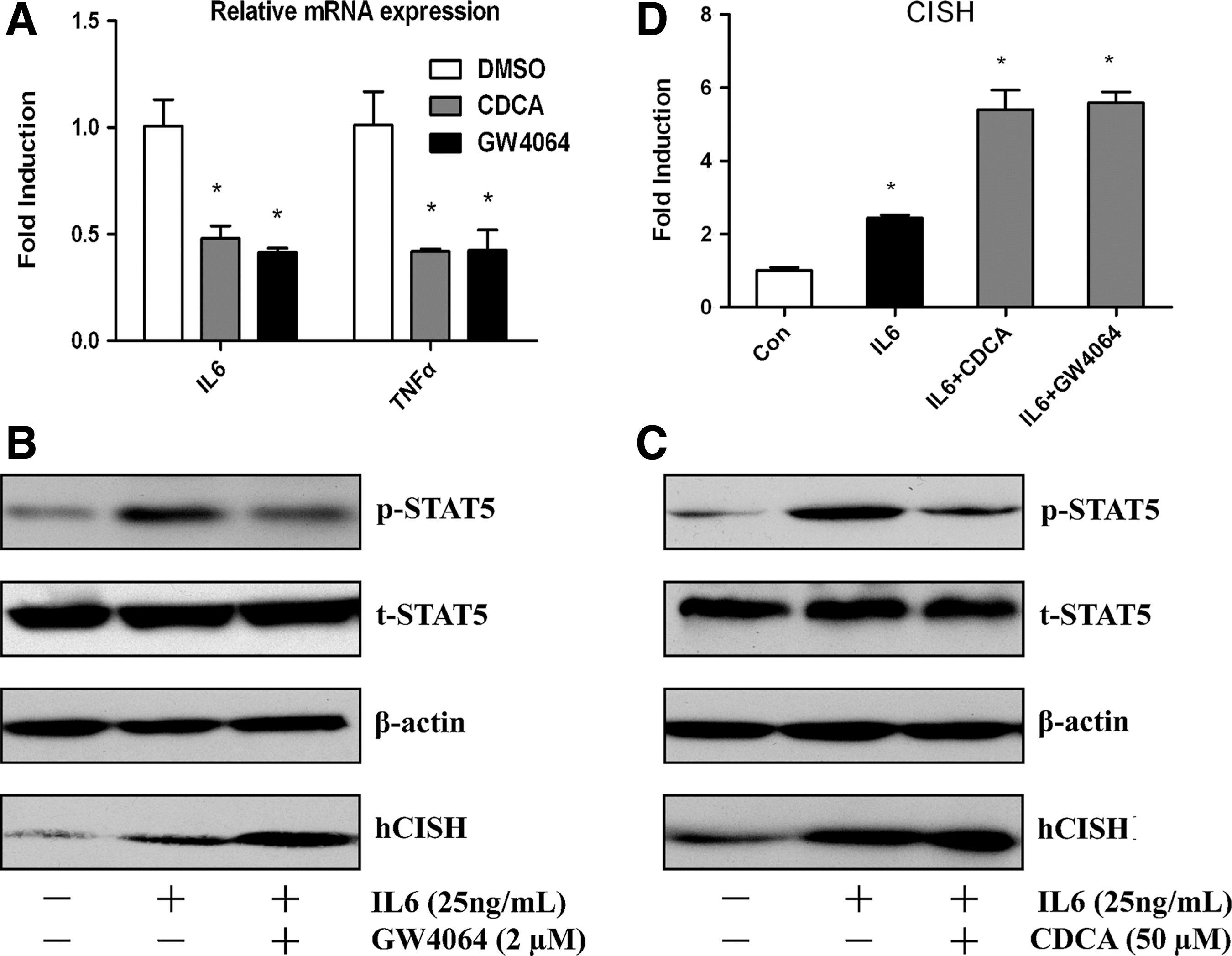
FXR ligands suppress pro-inflammatory cytokine expression and IL6-stimulated phosphorylation of STAT5.
FXR ligands enhance transcriptional activity of the human CISH promoter region
The above studies suggested CDCA and/or GW4064 enhanced CISH expression in vivo and in vitro. To further examine whether CISH promoter is a transcriptional target of FXR, we constructed a luciferase reporter expression plasmid (pGL3-CISH) that is driven by a human CISH promoter (−3009∼+199). Following transfection with pGL3-CISH, the HepG2 cells were treated with FXR ligands or vehicle. Due to enough endogenous FXR and retenoid X receptor (RXR) α in HepG2 cells, we did not transfect exogenous FXR and RXRα to the cells. Figure 4A indicated that enhancing the activity of the CISH promoter by CDCA and GW4064 was likely attributable to activation of endogenous FXR in HepG2 cells. Nonetheless, as shown in Fig. 4B, the promoter activity was increased when cells were co-transfected with an expression plasmid encoding a constitutively activated FXR [vpFXR was generated by fusing the VP16 activation domain to the N terminus of FXR cDNA and its constitutive activity was demonstrated using a tk-EcRE-Luc, an established FXR reporter gene (Xie and others 2001)], which clearly demonstrated that a genetic activation of FXR on CISH promoter activity.
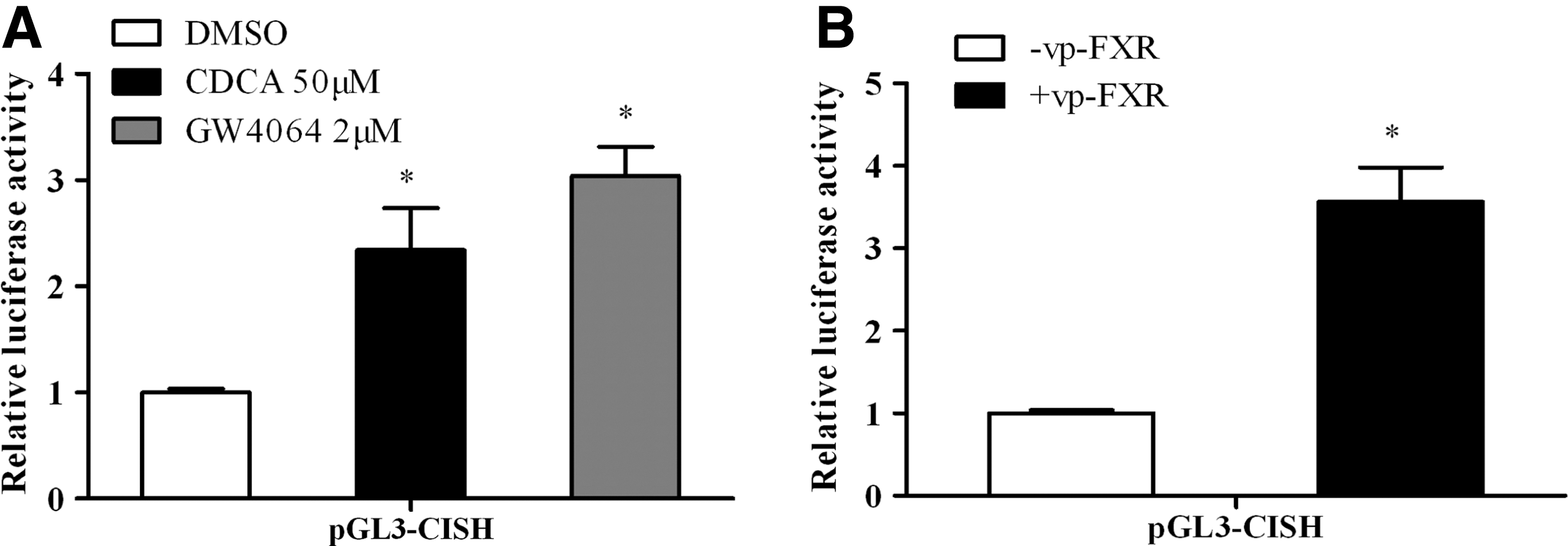
FXR induces the transcriptional activity of human CISH gene promoter.
Discussion
Cytokine-STAT5 signaling in hepatocyte regulates a wide range of hepatic genes, including metabolism enzymes and factors. CISH, the feedback modulator of STAT5 pathway, has classically been shown to be negative regulator of various cytokine signaling. And CISH has been demonstrated to limit the extent of Toll-like receptor signaling indirectly by inhibiting autocrine cytokine response (Baetz and others 2004). Moreover, there was a case report that variants of CISH were associated with susceptibility to infectious diseases caused by diverse pathogens, suggesting a possible role of CISH in the development of inflammatory and infectious diseases (Wang and Wang 2010).
Despite CISH being considered as an important negative regulator for inflammatory cytokine signaling, current understanding of the molecular basis underlying regulation of CISH expression is still very limited. Our studies revealed for the first time that the anti-inflammatory nuclear receptor FXR ligands can upregulate CISH expression in vivo and in vitro. Moreover, reporter assay demonstrated that CISH promoter activity was significantly increased via pharmacological or genetic activation of FXR. However, the mechanism by which FXR regulates CISH expression is not fully understood. A GenBank database search (
Strongly expressed in the liver and intestine, FXR has been shown to be the master transcriptional regulator of several enterohepatic metabolic pathways with relevance to the pathophysiology of conditions such as cholestasis, fatty liver disease, cholesterol gallstone disease, intestinal inflammation, and tumors. Besides its effects in anti-tumor and fibrosis, have been shown to suppress cellular transformation, proliferation, and invasion (Swales and others 2006; Kim and others 2007; Wang and others 2009), the potential therapeutic role of FXR agonists for the management of hepatic and intestinal inflammation is highlighted (Zhang and others 2009a, 2009b; Gadaleta and others 2011; Nijmeijer and others 2011). Moreover, there was a report (Vavassori and others 2009) that FXR−/− macrophages released, under basal conditions and in response to stimulation with lipopolysaccharide, increased amounts of pro-inflammatory cytokines compared with wild-type cells, indicating that FXR gene ablation results in a systemic dysregulation of pro-inflammatory gene expression. However, the precise mechanisms of action underlying the anti-inflammatory effects of FXR agonists are poorly understood.
Here, we report that FXR ligands could repress pro-inflammatory cytokine expression and attenuate IL6-induced STAT5 activation, which may be associated with upregulating CISH. Results from our study suggest that modulation of CISH expression may represent a novel mechanism through which FXR activation could selectively affect cytokine signaling. It remains to be determined whether the FXR-CISH pathway contributes to the prevention or initiation/progression of cytokine-related diseases. The answers to these questions might be found in studies in mice in which components of the FXR-CISH pathway are selectively modulated in those diseases.
Footnotes
Acknowledgment
This work was supported by the National Nature Science Foundation of China, No. 30771121 and No. 31071244.
Author Disclosure Statement
No competing financial interests exist.