
Abstract
The interferon regulatory factor (IRF)-3 transcription factor plays a central role in the capacity of the host to mount an efficient innate antiviral immune defense, mainly through the regulation of type I Interferon genes. A tight regulation of IRF-3 is crucial for an adapted intensity and duration of the response. Redox-dependent processes are now well known to regulate signaling cascades. Recent reports have revealed that signaling molecules upstream of IRF-3, including the mitochondrial antiviral-signalling protein (MAVS) and the TNF receptor associated factors (TRAFs) adaptors, are sensitive to redox regulation. In the present study, we assessed whether redox regulation of thiol residues contained in IRF-3, which are priviledged redox sensors, play a role in its regulation following Sendai virus infection, using a combination of mutation of Cysteine (Cys) residues into Alanine and thiols alkylation using N-ethyl maleimide. Alkylation of IRF-3 on Cys289 appears to destabilize IRF-3 dimer in vitro. However, a detailed analysis of IRF-3 phosphorylation, dimerization, nuclear accumulation, and induction of target gene promoter in vivo led us to conclude that IRF-3 specific, individual Cys residues redox status does not play an essential role in its activation in vivo.
Introduction
T
The ubiquitously expressed interferon regulatory factor (IRF)-3 transcription factor is central to the host innate immune antiviral response, through the transcriptional regulation of cytokines, mainly type I Interferon (IFN) β and IFN-α1 and the chemokine regulated upon activation, normal T cell expressed and secreted, but also IFN stimulated genes (ISGs), including IFIT1, 2, and 3 genes, encoding ISG56, ISG54, and ISG60 respectively [reviewed by Hiscott (2007)]. As a result of the activity of a CRM-1-dependent export from the nucleus, IRF-3 is constitutively present in the cytoplasm of resting cells (Kumar and others 2000). Activation of IRF-3 is initiated following the recognition of invading viruses by pattern recognition receptors, including members of the membrane-bound Toll-like receptors (TLRs) family, TLR-3, -7, and -9, and of the cytoplasmic retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs) family, RIG-I and melanoma-differentiation-associated gene 5 (MDA5) [reviewed by Kawai and Akira (2010) and Thompson and others (2011)]. Fine-tuned specific signaling cascades downstream of these receptors ultimately activate the IκB-kinase (IKK)-related kinases, TANK binding kinase-1 (TBK1) and IKKɛ, which phosphorylate IRF-3 at C-terminal phosphoacceptor sites, thereby inducing its dimerization and nuclear accumulation (Fitzgerald and others 2003; Sharma and others 2003). In the nucleus, activated IRF-3 associates with the histone acetyl transferase CREB-binding protein (CBP), or its related homologue p300, to form an active transcription complex that is able to bind to the IFN-stimulated response element present in the promoters of target genes [reviewed by Hiscott (2007)]. After genes transcription, IRF-3 is polyubiquitinylated leading to its degradation by the proteasome in the nucleus, thereby terminating its activation (Bibeau-Poirier and others 2006; Saitoh and others 2006). Post-translational modifications of IRF-3 by phosphorylation and ubiquitination are by far the best characterized, although modifications by sumoylation, acetylation, and glutathionylation have also been described (Suhara and others 2002; Kubota and others 2008; Prinarakis and others 2008).
Transiently increased reactive oxygen species (ROS) levels act as a cellular switch for signaling cascades that are important in various physiological processes, primarily through redox post-translational modification of proteins (Rhee 2006; Finkel 2011). Increasing data suggest the importance of redox-dependent regulation of the signaling pathway leading to IRF-3 activation (Arnoult and others 2011; West and others 2011). We recently showed that the expression of the mitochondrial antiviral-signalling protein (MAVS), which serves as a platform for the formation of the signalosome downstream of RLRs, is dependent on ROS produced by the NOX2 NADPH oxidase enzyme (Soucy-Faulkner and others 2010). Thiol groups in reactive Cysteine (Cys) residues in signaling molecules act as privileged redox sensors, allowing for protein's conformation or activity to be modulated (Rhee 2006; Finkel 2011). Glutathionylation of the TNF acceptor associated factor 3 (TRAF3), which is part of the MAVS-dependent signalosome upstream of IRF-3, was recently shown to be essential for its signaling capacity following infection by Herpes Simplex Virus (HSV)-1 (Gonzalez-Dosal and others 2011). Finally, IRF-3 itself was reported to be glutathionylated, a modification that is reversible by the Glutaredoxin 1 (Grx1) antioxidant enzyme (Prinarakis and others 2008).
The characterization of the redox sensitivity of key signaling proteins critical for the generation of the proinflammatory and antiviral responses provides an opportunity to develop new therapeutic strategies against viral conditions in which the host inflammatory response is deleterious. In the present study, we assessed the potential role of redox regulation of specific, individual thiol residues contained in IRF-3 on its activation during Sendai virus (SeV) infection through the mutation of IRF-3 Cys residues into Alanine (Ala) and reduced thiol alkylation using N-ethyl maleimide (NEM). Our data confirm that thiol groups contained in signaling molecules upstream of IRF-3 are essential for an efficient activation. However, although alkylation of IRF-3 Cys289 destabilizes IRF-3 dimer in vitro, IRF-3 specific, individual Cys redox status does not play an essential role in its activation in vivo.
Materials and Methods
Cells and viruses
A549 cells (ATCC) were cultured in F12K/Ham medium (Gibco) containing 10% heat-inactivated fetal bovine serum (HI-FBS) and 1%
Plasmids
The pRL-null Renilla reporter plasmid was obtained from Promega. The pCMVBL-flag, pCMVBL-flag-IRF-3, pcDNA3.1-zeo-myc-IRF-3 and pcDNA3.1-zeo-myc-IRF-3(5D) expression plasmids, and the IFIT1prom-pGL3 luciferase reporter plasmid were previously described (Lin and others 1998; Grandvaux and others 2002; Sharma and others 2003). pCMVBL-flag-IRF-3-C167A, C222A, C267A, C289A, C347A, C371A, and pcDNA3.1-zeo-myc-IRF-3(5D)-C289A encoding plasmids were generated through site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) following the manufacturer's protocol. All mutations were validated by sequencing and plasmids were selected for homogenous expression of IRF-3wt and the different IRF-3-C/A mutants (Fig. 1B
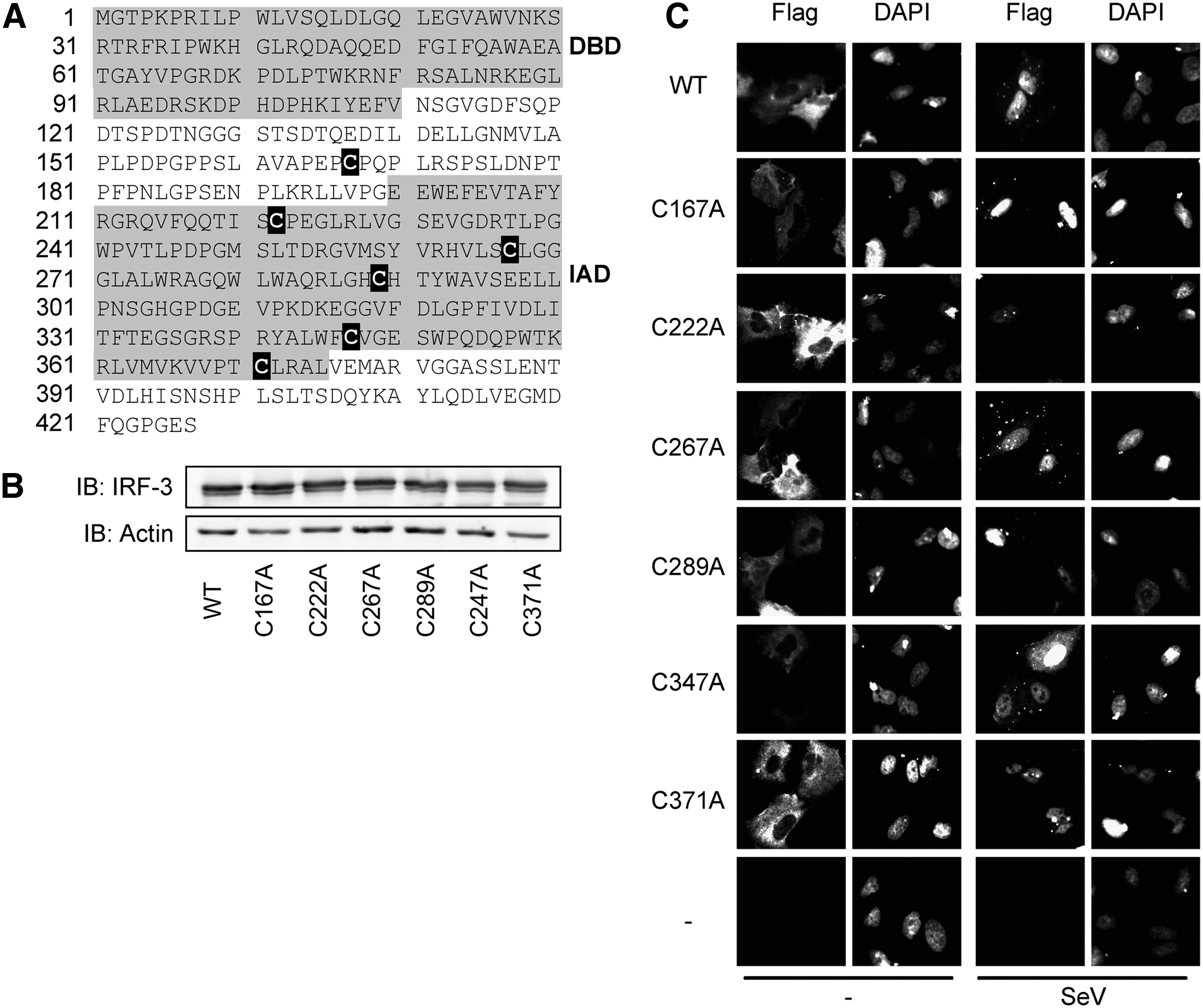
Mutation of individual interferon regulatory factor (IRF)-3 Cys into Ala does not impair its activation following Sendai virus (SeV) infection.

Infection and chemical treatments
Subconfluent A549 or MEF IRF-3−/− cells were infected with SeV at 5–100 hemagglutinating units (HAU)/106 cells in serum-free medium (SFM) for the first 2 h, after which 10% HI-FBS was added for the rest of the incubation period. When required, either NEM (Sigma-Aldrich) or the vehicle (ethanol) pretreatment was performed, at the indicated concentration, 1 h before infection in SFM. In the experiments where cells were transfected to express Myc-IRF-3wt, Myc-IRF-3(5D), or Myc-IRF-3(5D)-C289A, NEM treatment was performed for 1 h and then cells were washed and further cultured for another 4 h. In the experiments where NEM treatment was performed in vitro, the incubation of whole cell extracts (WCE) with NEM or ethanol (vehicle) was performed on ice for 15 min prior to further analysis.
Immunoblot analysis
WCE were prepared in Nonidet P-40 lysis buffer, as previously described (Solis and others 2007), and quantified using the Bio-Rad Protein Assay (Bio-Rad). WCE were subjected to sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) electrophoresis and analyzed by immunoblot using the anti-IRF-3-Serine 396 phosphospecific (IRF-3-P-S396; Servant and others 2003), anti-IRF-3 (Active Motif), anti-actin (Chemicon International), or anti-Flag M2 (Sigma-Aldrich). In between phosphospecific- and anti-IRF-3-antibodies, the membrane was stripped in 0.2% SDS, 62.5 mM Tris-HCl pH 6.8, 0.1 mM β-mercaptoethanol for 20 min at 50°C. Immunoreactive bands were visualized by enhanced chemiluminescence using the Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences) acquired on an ImageQuant LAS 4000mini apparatus (GE Healthcare Life Sciences).
IRF-3 dimerization assay
Native-PAGE were conducted as described (Iwamura and others 2001) using 10 μg WCE and 7.5% acrylamide gel. After transfer into nitrocellulose membrane, monomeric and dimeric forms of IRF-3 were detected using the immunoblot protocol described above using anti-IRF-3, anti-Flag M2, or anti-Myc antibodies.
Indirect immunofluorescence
A549 cells were grown on coverslips. After the indicated treatment and/or infection, cells were fixed in 3% paraformaldehyde in phosphate-buffered saline (PBS) containing 0.5 mM CaCl2 and 0.5 mM MgCl2 (PBS/Ca/Mg) for 15 min at room temperature (RT), then permeabilized in 0.5% triton in PBS/Ca/Mg for 15 min at RT, and finally blocked in PBS/Ca/Mg containing 10% goat serum for 1 h at RT and incubated with anti-flag M2 (1/100) in PBS/Ca/Mg containing 3% bovine serum albumin for 2 h at RT. Thereafter, the cells were incubated with AlexaFluor 568-labeled goat anti-mouse (1/1,000; Invitrogen) for 1 h in the dark at RT. Nuclei counterstaining was performed by incubation with 4′,6-diamidino-2-phenylindole (1 μg/mL) for 5 min in the dark at RT. Between every steps, cells were washed thrice in PBS/Ca/Mg. Coverslips were mounted on slides using gel-mount (Biomeda). Fluorescence was visualized using a LEICA DM6000 microscope. Pictures were analyzed with Openlab 4.0.4 software (Improvision).
Luciferase reporter assays
MEF IRF-3−/− cells in 24-well plates were cotransfected with the pRL-null Renilla (Renilla luciferase, internal control, 50 ng), the IFIT1prom-pGL3 luciferase reporter (firefly luciferase, 100 ng) constructs, the empty expression plasmid (300 ng), and the indicated IRF-3 expression plasmid (50 ng) using the TransIT-LT1 transfection reagent (Mirus). Where indicated, SeV infection was performed 16 h post-transfection at 100 HAU/106 cells. Quantification of luciferase was performed 24 h post-infection using the dual luciferase reporter assay kit (Promega) according to the manufacturer's instructions. In experiments where MEF IRF-3−/− cells were transfected to express Myc-IRF-3wt, Myc-IRF-3(5D), or Myc-IRF-3(5D)-C289A, luciferase activities were quantified 24 h post-transfection. Relative luciferase activities were calculated as the luciferase to Renilla ratio.
Results
Mutation of IRF-3 Cys into Ala does not impair its activation following SeV infection
To start investigate the regulation of IRF-3 through its potentially redox reactive thiols, the six Cys of IRF-3, Cys167, Cys222, Cys267, Cys289, Cys347, and Cys371 (Fig. 1A), were individually mutated into Ala (IRF-3-C/A) to suppress their capacity to be oxidized. MEF IRF-3−/− cells were individually transfected with plasmids encoding either Flag-IRF-3wt or each of the Flag-IRF-3-C/A mutants to verify homogenous expression (Fig. 1B). IRF-3 activation status was first analyzed in A549 cells through the analysis of its subcellular localization by indirect immunofluorescence both at basal level and following SeV infection (100 HAU/106 cells for 6 h). As shown in Fig. 1C, IRF-3wt and each of the IRF-3-C/A mutants exhibited similar cytoplasmic expression at basal level and translocated into the nucleus after SeV stimulation. Next, the capacity of IRF-3wt and IRF-3-C/A mutants to transactivate the IRF-3-dependent IFIT1 promoter (Grandvaux and others 2002) after SeV infection was assessed by luciferase reporter assay. To exclude the activity of endogenous IRF-3, these experiments were performed in MEF IRF-3−/− cells. As shown in Fig. 1D, each of the IRF-3-C/A mutants ectopically expressed in MEF IRF-3−/− cells was able to reconstitute an SeV-inducible activation of the IFIT1 promoter comparable to that mediated by IRF-3wt. Altogether, these results demonstrate that impairment of the capacity of IRF-3 to be oxidized on specific, individual Cys residues does not alter its capacity to be activated and to transactivate target gene promoter following SeV infection.
Alkylation of thiol groups using NEM impairs IRF-3 activation in vivo and destabilizes IRF-3 dimer in vitro
To further understand the role of thiols in the redox regulation of IRF-3, we studied the impact of thiol alkylation using NEM during SeV infection of A549 cells. NEM has been extensively used to probe the functional thiol groups in a variety of signaling proteins, including transcription factors (Bhanoori and others 2003). First, we assessed whether NEM treatment prior to SeV infection affected endogenous IRF-3 phosphorylation and dimerization, two critical steps in its activation. As shown in Fig. 2A, SeV-induced phosphorylation of IRF-3 at Ser396 was significantly reduced in cells pretreated with NEM at 25 μM compared with vehicle-treated cells. Additionally, the formation of the active dimeric form of IRF-3, evaluated by native-PAGE, was also effectively impaired in a dose-dependent manner (Fig. 2B). Based on data from the literature (Gonzalez-Dosal and others 2011), molecules in the upstream cascade leading to IRF-3 activation could be targeted by alkylation. Thus, to evaluate whether NEM could target IRF-3 itself, NEM-mediated alkylation of WCE derived from A549 either left uninfected or infected with SeV was performed in vitro followed by analysis of IRF-3 phosphorylation and dimerization. As expected, since phosphorylation is a covalent modification that occurs before the treatment, NEM treatment did not affect IRF-3 Ser396 phosphorylation (Fig. 3A). However, NEM treatment led to the disruption of IRF-3 dimer as observed by the recovery of IRF-3 in its monomeric form (Fig. 3B). Altogether, these data demonstrate that alkylation impairs IRF-3 activation in vivo. Moreover, IRF-3 possesses Cys residue(s) that is (are) potential target(s) for alkylation and that is (are) essential for the stability of the dimer.
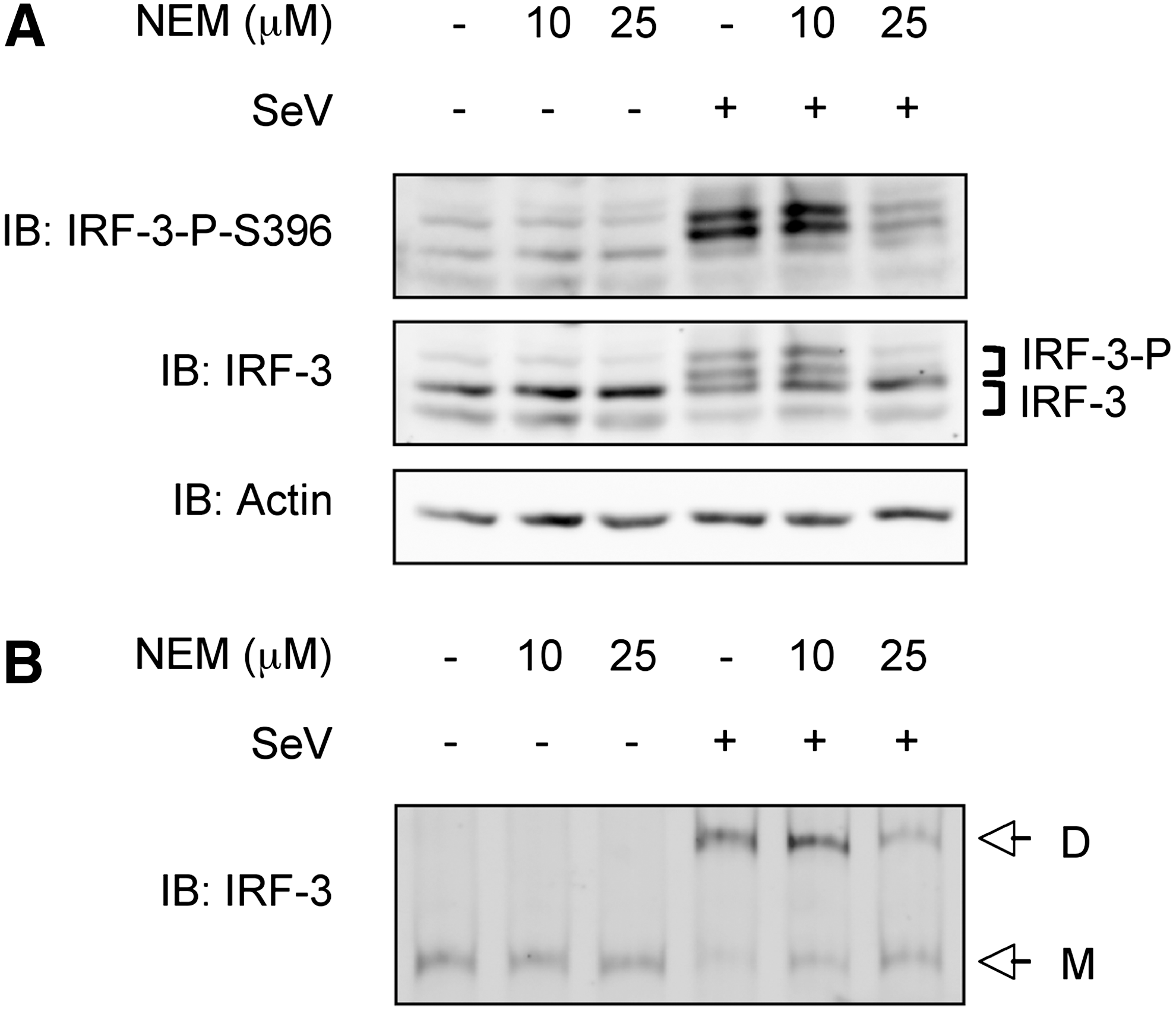
Alkylation of thiols impedes IRF-3 activation in vivo. A549 cells were pretreated with the indicated concentration of N-ethyl maleimide (NEM) or ethanol (vehicle) for 1 h and left uninfected or infected with SeV (100 HAU/106 cells) for 4 h.
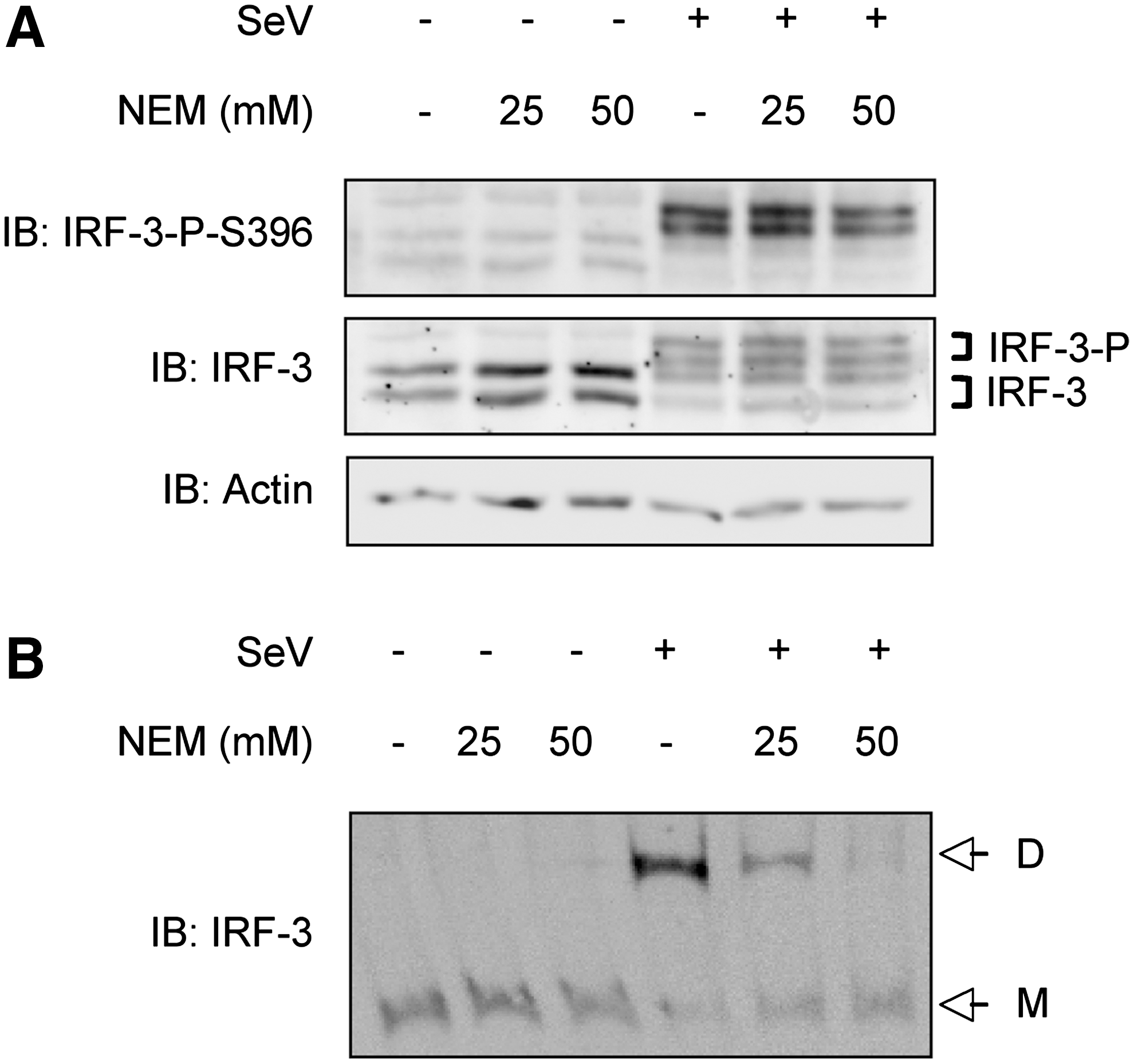
Alkylation of thiols destabilizes IRF-3 dimer in vitro. WCE from A549 uninfected or infected with SeV (40 HAU/106 cells) for 6 h, were treated with the indicated concentration of NEM or ethanol (vehicle) for 15 min. Following treatment, WCE were analyzed by SDS-PAGE
Mutation of Cys289 into Ala prevents in vitro disruption of IRF-3 dimers by NEM
As the IRF-3 dimer is disrupted following NEM treatment in vitro, suggesting that IRF-3 could be directly alkylated, IRF-3-C/A mutants were further used to characterize the potential site of IRF-3 alkylation. WCE were prepared from A549 individually transfected with Flag-IRF-3wt or Flag-IRF-3C/A mutants expressing constructs and either left uninfected or infected with SeV at 40 HAU/106 cells for 6 h. WCE were further treated with NEM in vitro. As shown in Fig. 4, IRF-3wt and each of the IRF-3-C/A mutants were able to form dimers upon SeV stimulation. Similar to IRF-3wt, dimers formed from the IRF-3-C167A, IRF-3-C222A, IRF-3-C267A, IRF-3-C347A, and IRF-3-C371A exhibited sensitivity to NEM (Fig. 4A, C). Interestingly, dimers containing IRF-3-C289A were resistant to NEM treatment. Similar experiment performed with WCE generated from reconstituted MEF IRF-3−/− led to the same observations (Fig. 4B, C). Based on these results, IRF-3 Cys289 is targeted by NEM and its alkylation destabilizes IRF-3 dimer in vitro.
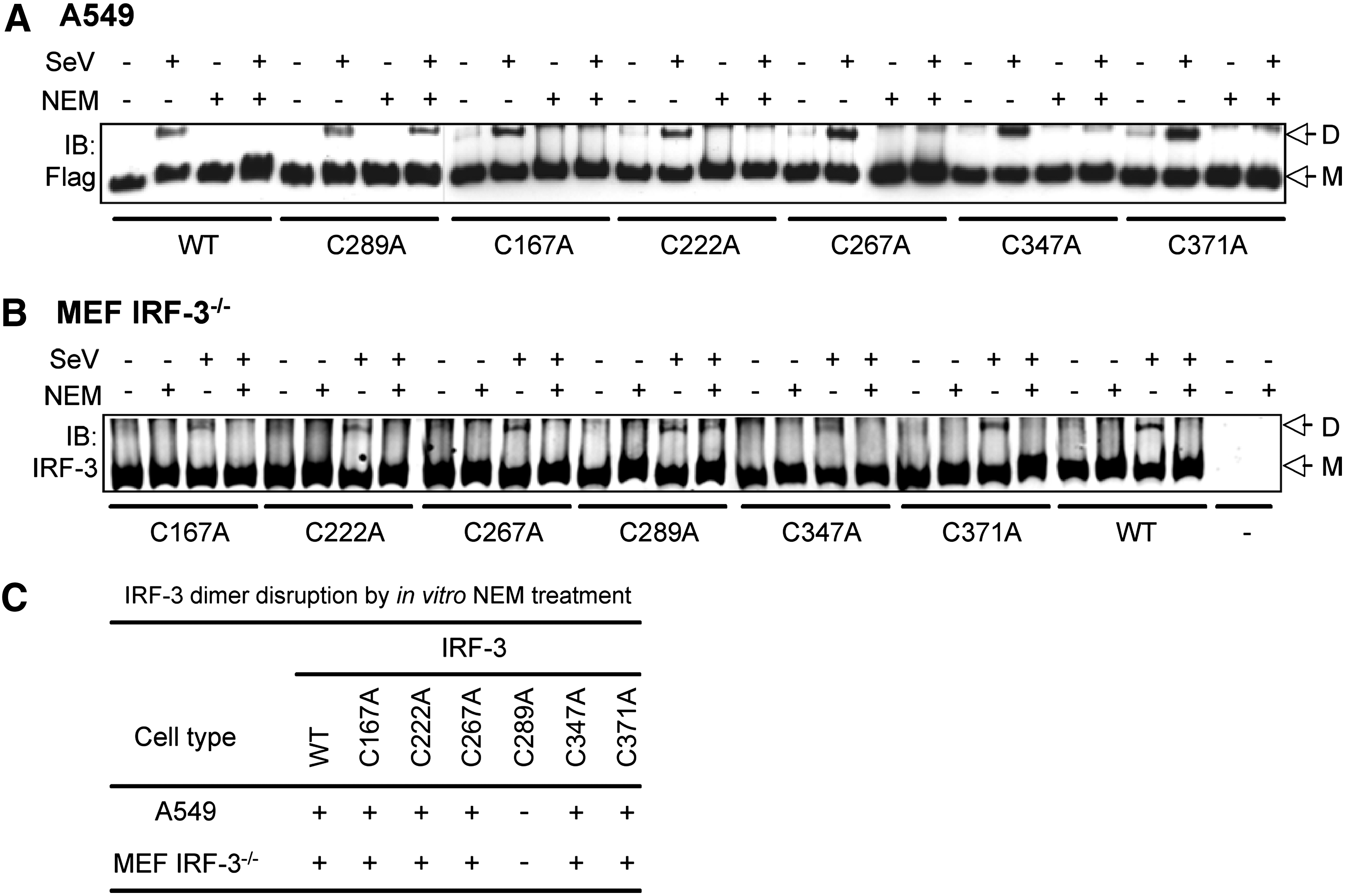
IRF-3 Cys289 mutation into Ala prevents disruption of IRF-3 dimer by NEM in vitro. A549
Mutation of Cys289 into Ala does not affect IRF-3 dimerization in vivo
Based on the in vitro experiments, the role of IRF-3 Cys289 alkylation in IRF-3 dimerization was further studied in vivo, despite the lack of a phenotype of its mutation in the capacity of IRF-3 to translocate to the nucleus, activate target promoter, and mount an antiviral response (Fig. 1). A detailed kinetic of SeV-induced IRF-3wt and IRF-3-C289A dimerization was monitored in transfected A549 (Fig. 5A) and MEF IRF-3−/− (Fig. 5B) cells. As observed in Fig. 5A and B, the kinetic and intensity of IRF-3 dimer formation was similar in both conditions. In addition, in A549 cells, SeV-induced dimers containing IRF-3-C289A were sensitive to NEM pretreatment similar to IRF3wt (Fig. 5C). Contrary to the in vitro experiments, the observed effect of NEM treatment in vivo does not seem to be mediated by the direct alkylation of IRF-3. To confirm this result, the constitutively active IRF-3(5D) phosphomimetic mutant (Lin and others 1998) and the IRF-3(5D)-C289A mutant were used to analyze IRF-3 dimerization independent of the upstream signaling cascade in A549 cells. As shown in Fig. 5D, in vivo NEM treatment was not able to disrupt IRF-3(5D)-containing dimers. Additionally, IRF-3(5D)-C289A mutant retained the capacity to constitutively form dimers and was not affected by NEM treatment. Further analysis of IRF-3 transcriptional activity using IFIT1 promoter as a reporter, revealed that IRF-3(5D) and IRF-3(5D)-C289A similarly induced IFIT1 promoter activity when ectopically expressed to reconstitute MEF IRF-3−/− cells (Fig. 5E
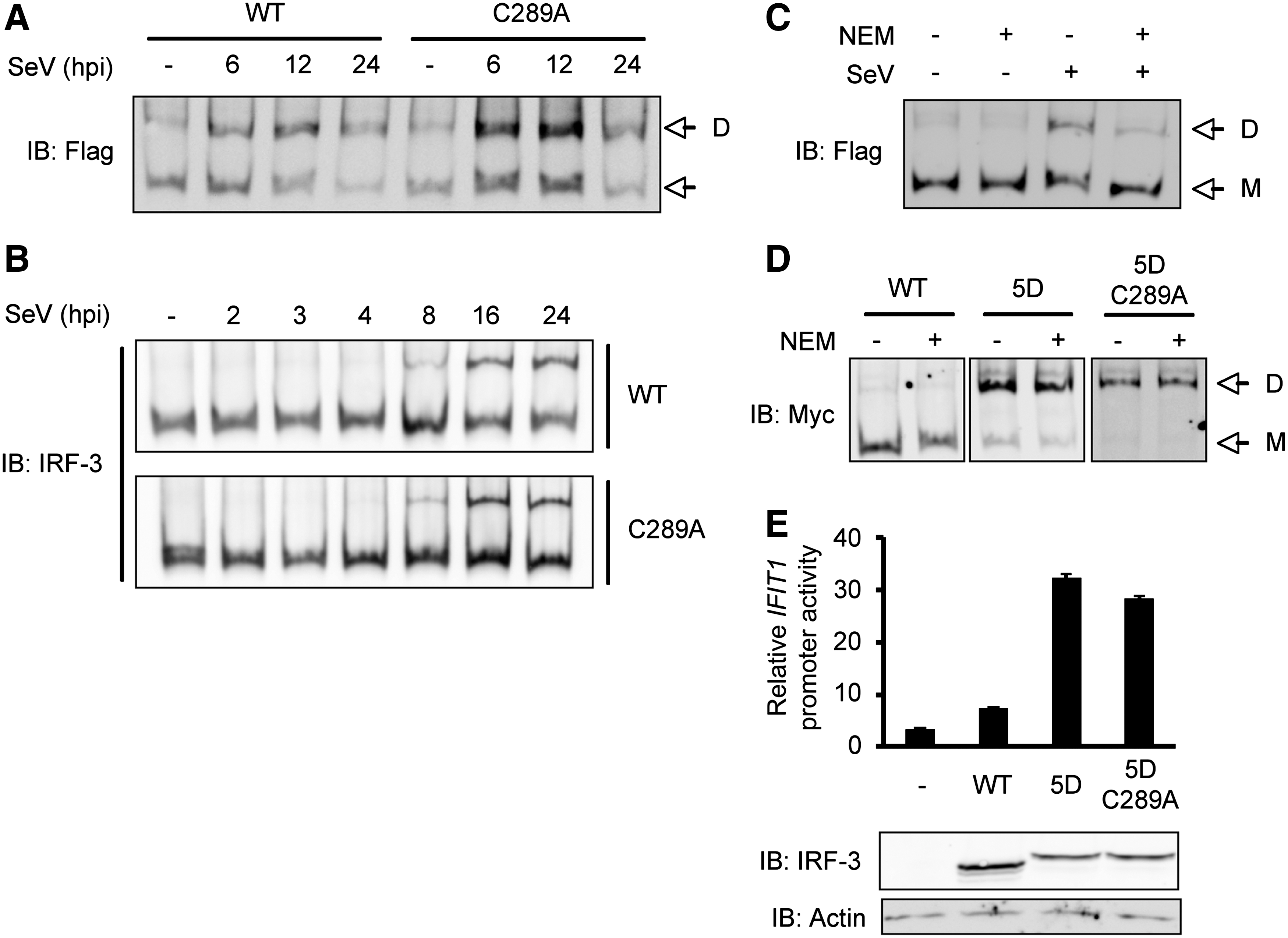
Mutation of Cys289 into Ala does not affect IRF-3 dimerization in vivo. A549
Discussion
In this study, we investigated the role of IRF-3 specific, individual thiol as redox-sensitive sensors implicated in IRF-3 activation. Cys residues in signaling molecules act as privileged redox sensors, which can be modified in several ways, including formation of inter- or intra-molecular disulfide bonds, S-nytrosylation, direct oxidation to sulfenic acid or to higher order sulfinic or sulfonic acid, and formation of mixed disulfides with glutathione (Ghezzi and Bonetto 2003; Biswas and others 2006). If thiols are essential, it would be expected that blockade of thiol residues through their alkylation, using thiol-alkylating agent NEM, would affect SeV-induced IRF-3 activation, as published for other signaling pathways (Bhanoori and others 2003). The observation that the treatment of cells with NEM before SeV infection led to inhibition of IRF-3 activation, measured by inhibition of its phosphorylation and dimerization (Fig. 2), is in line with the recent observations that upstream signaling molecules, such as TRAF3, are subjected to regulation by glutathionylation (Gonzalez-Dosal and others 2011). We also recently showed that ROS derived from the NOX2 NADPH oxidase are essential for airway epithelial cells to trigger an efficient IRF-3 activation through the regulation of MAVS expression level (Soucy-Faulkner and others 2010). However, whether this pathway involves thiol oxidation remains to be determined. In the present study, we focused on IRF-3 thiol function in its regulation. We found that IRF-3-C/A mutants are activated similar to IRF-3wt following SeV infection in A549 or reconstituted MEF IRF-3−/- cells based on their capacity to translocate in the nucleus, dimerize, and transactivate target gene promoter (Fig. 1). These results indicate that the incapacity of IRF-3 individual thiol residues to become oxidized during infection does not impair its activation.
In vitro alkylation of Cys289 led to IRF-3 dimer dissociation (Figs. 3 and 4), suggesting a potential role of Cys289 redox status in IRF-3 dimer stability. However, SeV-induced dimers containing IRF-3-C289A remain sensitive to NEM in vivo, similarly to IRF-3wt (Fig. 5). This suggests that IRF-3 alkylation on Cys289A either does not occur or does not affect IRF-3 dimerization in vivo. A limitation of this approach is that we cannot exclude that the effect of NEM treatment in vivo on IRF-3 itself is masked by the alkylation of upstream signaling molecules. Answering this question would require the means to perform specific IRF-3 alkylation. This strategy was recently used to demonstrate the role of the alkylation of Cys468 of the STAT3 transcription factor in the inhibition of its nuclear accumulation (Buettner and others 2011). In this study, the authors used a chemical compound, C48 that specifically alkylates Cys468 of STAT3. A strategy aimed at identifying such a compound to specifically alkylate IRF-3 Cys289 would help validate our in vitro observation. Alignment of the human IRF-3 primary sequence with murine and rat sequences (data not shown) revealed that Cys289 is not conserved between species, contrary to Cys222, Cys347 and Cys371, which argues against an essential role of Cys289 in IRF-3 regulation. Similarly, an analysis of the available crystal structure of IRF-3 revealed that Cys289, which is comprised in the IRF association domain (IAD) that mediates homo- and heterodimeric interactions, is not located at the direct interface between monomers (Takahasi and others 2003). Thus, it is unlikely that alkylation of Cys289 might sterically affect the stability of the dimer. However, it cannot be excluded that alkylation introduces structural perturbations that hinder the dimeric association as it has been suggested for glutathionylation of p53 (Velu and others 2007). The observation that dimers formed from the constitutively active form of IRF-3, IRF-3(5D), or the IRF-3(5D) containing Cys289 mutated into Ala, IRF-3(5D)-C289A, are resistant to NEM treatment in vivo (Fig. 5) also argues that in the absence of upstream signaling cascade, thiol alkylation does not affect IRF-3 dimerization.
It was reported that IRF-3 is subjected to S-glutathionylation on an uncharacterized residue in HEK293 and HeLa-transfected cells. This modification appears to occur at basal level and was subjected to deglutathionylation by the Grx1 enzyme following SeV infection (Prinarakis and others 2008). However, we failed to detect endogenous IRF-3 glutathionylation in A549 cells using the anti-GSH antibody (data not shown). Further, IRF-3 Cys289 can be alkylated by NEM in vitro (Fig 4), demonstrating that Cys289 thiol group is available for such alkylation and is thereby not gluthationylated following SeV infection. A lack of IRF-3 glutathionylation detection was also recently reported in a study performed in primary murine peritoneal macrophages (Gonzalez-Dosal and others 2011). Since redox regulation of signaling pathways and transcription factors is now well documented to be cell-type specific, as illustrated by the well-documented redox regulation of the NF-κB activation pathway (Grandvaux 2011), it cannot be excluded that glutathionylation of IRF-3 is a cell-type specific mechanism.
In conclusion, according to the present study, we conclude that the redox status of the individual thiol residues of IRF-3 does not impact on its activation and capacity to control SeV replication. Further studies are required to evaluate the possibility that IRF-3 may be targeted by other oxidative modifications on alternative residues, such as carbonylation. Considering the redox regulation of the upstream signaling cascade leading to IRF-3 activation, it would be of interest to identify new targets for thiol modification in this cascade that could be used in therapeutic strategies to resolve virus-related diseases.
Footnotes
Acknowledgments
The authors thank the members of the laboratory for helpful discussions and technical help. The authors also wish to thank Anton Soucy-Faulkner for preliminary data and production of IRF-3-C/A mutants and Dr. K. Mossman (McMaster University, Hamilton, Canada) for providing MEF IRF3−/− cells used in this study. We also thank Dr. C. Rose (CRCHUM, Montréal, Canada) for critical reading of the article. The present work was funded by an operating grant from the Canadian Institutes of Health Research (CIHR, Canada) #MOP89807 to N.G. and funds from the Fonds de la Recherche en Santé du Québec (FRSQ, Canada). N.Z. was recipient of a postdoctoral fellowship from the FRSQ/Institut National de la Santé et de la Recherche Médicale (INSERM, France). N.G. was recipient of a Tier II Canada Research Chair.
Author Disclosure Statement
No competing financial interests exist.