
Abstract
HIV is able to outpace the innate immune response, including that mediated by interferon (IFN), to establish a productive infection. Primary macrophages, however, may be protected from HIV infection by treatment with type I IFN before virus exposure. The ability of HIV to modulate the type I IFN-mediated innate immune response when it encounters a cell that has already been exposed to IFN remains poorly defined. The optimal pretreatment time (12 h) and the most potent HIV-inhibitors (e.g., IFN-α2 and -ω) were identified to investigate the ability of HIV to modulate an established type I IFN response. Gene expression at the level of the entire transcriptome was then compared between primary macrophages treated with type I IFNs, as opposed to treated with IFNs and then infected with HIV. Although HIV was not able to establish a robust infection, the virus was able to downregulate a number of IFN-stimulated genes (ISGs) with a fold change greater than 1.5 (i.e., AXL, IFI27, IFI44, IFI44L, ISG15, OAS1, OAS3, and XAF1). The downregulation of OAS1 by the presence of HIV was confirmed by real-time quantitative polymerase chain reaction. In conclusion, even though HIV replication is significantly inhibited by IFN pretreatment, the virus is able to downregulate the transcription of known antiviral ISGs (e.g., IFI44, ISG15, and OAS1).
I
A small number of ISGs whose protein products contribute to the inhibition of HIV replication have been identified and include apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G (APOBEC3G) (Peng and others 2006), bone marrow stromal cell antigen 2 (BST2) (Van Damme and others 2008), 2′-5′-oligoadenylate synthetase 1 (OAS1) (Schroder and others 1990), eukaryotic translation initiation factor 2-alpha kinase 2 (EIF2AK2, also known as PKR) (Muto and others 1999), and interferon-stimulated gene 15 (ISG15) (Kunzi and others 1995). APOBEC3G can inactivate HIV by deamination of viral cDNA, but this enzyme is targeted for proteosomal degradation by the HIV Vif protein (Sheehy and others 2003; Yu and others 2003). Similarly, BST2 restricts virion release from the surface of HIV-infected cells, but is counteracted by the Vpu protein of HIV (Van Damme and others 2008). EIF2AK2 inhibits HIV replication by inactivating the eukaryotic translation initiation factor 2A (eIF2A), which leads to broad-spectrum inhibition of protein synthesis, while genes of the OAS family activate RNaseL to cleave viral ssRNA (Stark and others 1998). Finally, ISG15 leads to the retention of HIV unspliced transcripts in the nucleus preventing their migration to the cytoplasm and subsequent translation (Kunzi and others 1995).
After HIV infection, viral replication outpaces the potentially protective innate immune responses mediated by IFN (Woelk and others 2004). What is less clear is what happens when HIV encounters a cell that has already been exposed to IFN. We hypothesized that in addition to countermeasures at the protein level against antiviral ISGs (e.g., Vif/APOBEC3G and Vpu/BST2), HIV may also circumvent innate immune responses by downregulating ISG expression at the transcriptional level. To address this hypothesis, microarray gene expression analysis was performed in primary monocyte-derived macrophages (MDMs) that were treated with IFN for comparison to MDMs treated with IFN, and then infected with HIV. Our previous work using this experimental design along an acute time course demonstrated that TNF receptor-associated factor 6 (TRAF6) was upregulated 8 h after an 18-h treatment of IFN-α2, but then downregulated when HIV was present (Sirois and others 2011). TRAF6 is a known mediator of toll-like receptor signaling and activates interferon regulatory factor 7 (IRF7), and nuclear factor kappa-B (NF-κB) (Konno and others 2009). To assess the effects of HIV on downstream ISGs regulated by IFN signaling, the current work focused on a later time course following infection (Days 1, 4, and 8) and was expanded to evaluate the HIV inhibitory properties of 6 IFN species from 4 different classes (IFN-α1, -α2, -α7, -β, -τ4, and -ω).
MDMs were selected for analysis because they represent a persistent viral reservoir (Wahl and others 2003) and demonstrate a robust phenotype of HIV inhibition following IFN pretreatment (Yamada and others 1988; Gendelman and others 1990; Meylan and others 1993). MDMs were obtained from 3 different donors as previously described (Woelk and others 2004). Briefly, monocytes were isolated by adherence from the peripheral blood mononuclear cells (PBMCs) of healthy donors and cultured over 5 days in 48-well plates in the presence of macrophage colony-stimulating factor (50 ng/mL) at a density that gave rise to 2×105 MDMs per well. Multiple wells were seeded per condition to provide replicates for statistical assessment. Type I IFNs were obtained from PBL Laboratories (Piscataway, NJ) and used in single pretreatments of MDMs at concentrations of 186 pM, which is equivalent to 1,000 IU of IFN-α2. Infection of MDMs with HIV was performed using the BaL strain at a multiplicity of infection (MOI) of 0.3 and unabsorbed virus was removed after 2 h by washing. HIV replication was assessed using an enzyme-linked immunosorbent assay (ELISA) for p24 core antigen (PerkinElmer, Waltham, MA). MDMs were lysed in 1 mL TRIzol (Invitrogen Life Technologies, Carlsbad, CA) for total RNA extraction to perform analyses of gene expression. Microarray analysis of the entire transcriptome was performed as previously described (Woelk and others 2010) using the HumanWG-6 v3 BeadChip from Illumina (San Diego, CA). Briefly, raw gene expression data were subjected to quantile normalization following log2 transformation, and quality control analysis indicated that there were no outlier arrays. Microarray gene expression data were deposited in the Gene Expression Omnibus (
To determine the optimal pretreatment time required to protect MDMs from HIV infection, MDMs were treated with 186 pM of IFN-α2 for 0, 2, 6, 12, and 18 h, and then infected with HIV (MOI of 0.3). HIV replication was assessed using an ELISA for p24 at days 1, 4, and 8 following infection (Supplementary Fig. S1; Supplementary Data are available online at
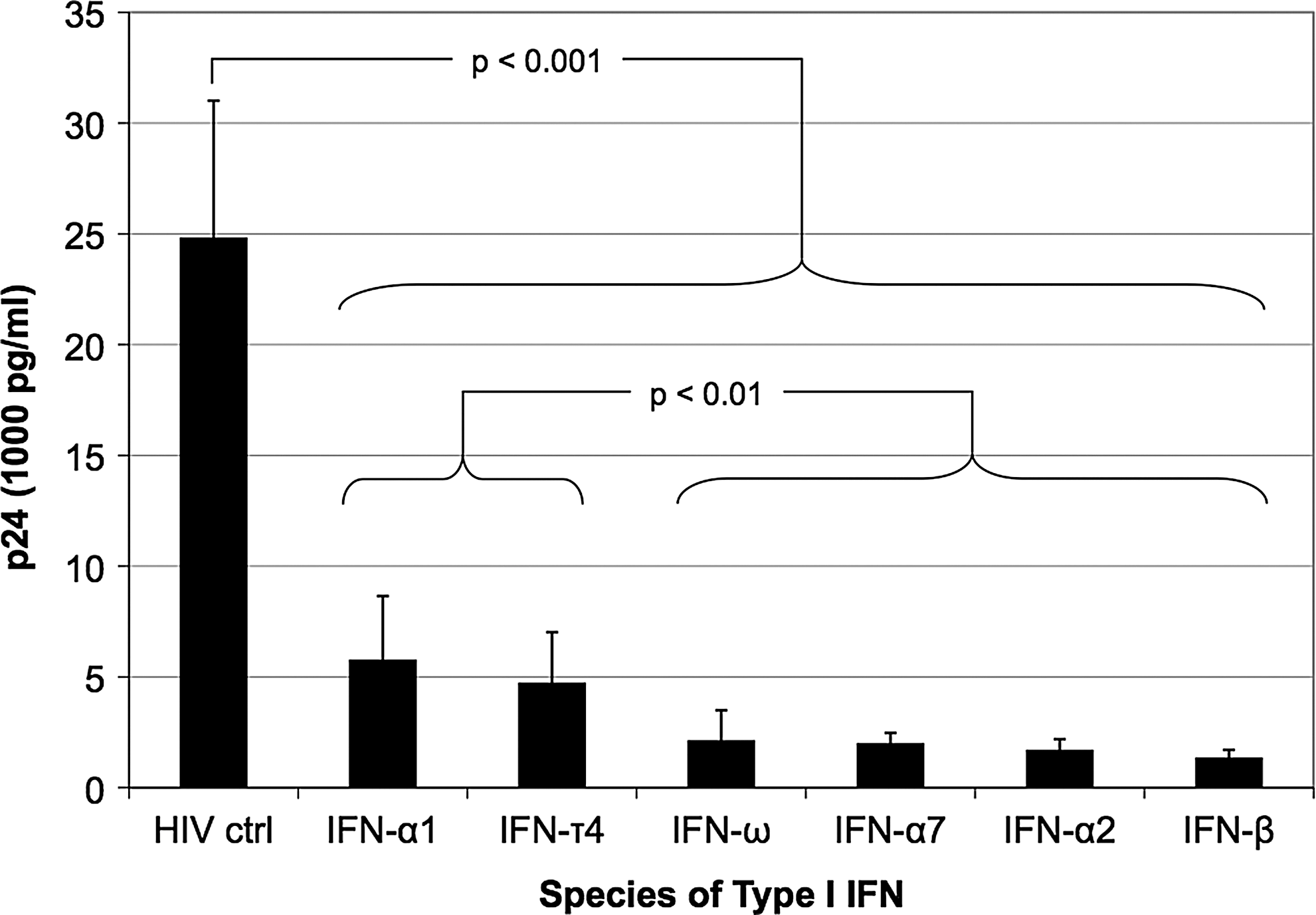
Different species of Type I interferon (IFN) vary in their ability to inhibit HIV replication. Monocyte-derived macrophages (MDMs) from a single donor (Donor 1) were pretreated for 12 h with 6 different species (4 different classes) of Type I IFN (186 pM), and then infected with HIV BaL [multiplicity of infection (MOI)=0.3] for 2 h. HIV replication was measured by an enzyme-linked immunosorbent assay (ELISA) for p24 antigen on day 8 after HIV infection. Each data point represents an average p24 measurement derived from 6 replicate wells. Statistical comparisons were performed using a one-way analysis of variance with a Tukey post hoc test. Error bars reflect the standard deviation of the mean.
To investigate the presence of HIV on an established IFN response, MDMs were subjected to 4 different conditions: (1) IFN-treated only, (2) IFN-treated followed by HIV infection, (3) HIV-infected only, and (4) a mock-treated and mock-infected control. Microarray gene expression analysis was performed on a total of 24 samples derived from the 4 conditions assessed at 3 time points (1, 4, and 8 days following treatment/infection) for both IFN-α2 or -ω. Microarray data were subjected to unsupervised clustering, which clusters samples based on the expression of genes in each sample, revealing the underlying biological structure in the data. This analysis revealed that all samples at day 1 clustered together, but samples from days 4 and 8 were intermingled based on experimental conditions (Supplementary Fig. S2). This suggests that experimental conditions were not exerting noticeable effects on gene expression at Day 1. Therefore, the gene expression data at Days 4 and 8 were interrogated further to determine the effects of HIV on an established IFN response. Fold change ratios were calculated for each gene by comparing each condition to the control, and then passed through a series of filters. Initially, ISGs were identified as those that were upregulated greater than 2-fold by IFN alone (condition 1) at both Days 4 and 8. Then, the IFN-treated condition was compared to the IFN-treated followed by the HIV infection condition to identify those ISGs that were downregulated at least 1.5-fold by the presence of HIV at both days. Assuming that it would be counterproductive for HIV infection by itself to induce the expression of ISGs with putative anti-HIV effects, those ISGs that were upregulated greater than 2-fold in the HIV control were removed. Finally, ISGs that passed these filters and were concordant with both IFN-treatments (IFN-α2 and -ω) were identified and corresponded to the following 8 ISGs: AXL receptor tyrosine kinase (AXL), interferon alpha-inducible protein 27 (IFI27), interferon-induced protein 44 (IFI44), interferon-induced protein 44-like (IFI44L), ISG15, OAS1, OAS3, and XIAP-associated factor 1 (XAF1) (Fig. 2). No genes were found to be downregulated by IFN-treatment and subsequently upregulated by the presence of HIV.

IFN stimulated genes downregulated by HIV. A heatmap depicting the 8 genes that were upregulated by IFN, downregulated by the presence of HIV and not upregulated by the virus itself, consistently across time points (Day 4 and 8) and IFN treatments (IFN-α2 and -ω). MDMs from a single donor (Donor 2) were subjected to 4 experimental conditions: (1) IFN-treatment (IFNA2 or IFNW), (2) IFN-treatment followed by HIV-infection (IFNA2+HIV or IFNW+HIV), (3) HIV-infection only (HIV), and (4) mock-treatment and mock-infection (MDM control). IFN pretreatments were for 12 h at a concentration of 186 pM, and HIV BaL infections were for 2 h at an MOI of 0.3. Fold changes were calculated between the experimental conditions and the MDM control, and then log2 transformed. For visualization purposes, log2 fold changes across conditions were median centered for each gene as reflected in the scale bar at the bottom left corner of the heatmap. Hierarchical clustering of genes was performed by calculating distances using the Pearson correlation metric, which were then clustered using the average linkage method.
The expression of OAS1 was validated by RT-qPCR for all time points (1, 4, and 8 days) for each of the 4 conditions for the experiment with IFN-α2, confirming our microarray gene expression findings. Fold changes were normalized to the expression of OAS1 in the MDM nontreated and noninfected control at day 1 and revealed that this gene was indeed considerably upregulated by IFN-α2, downregulated by the presence HIV, but not upregulated by HIV infection alone (Fig. 3A). Furthermore, an ELISA for p24 antigen was performed at day 1, 4, and 8 postinfection and confirmed that the downregulation of OAS1 by HIV following IFN treatment occurred despite the fact that viral replication was significantly inhibited (Fig. 3B).
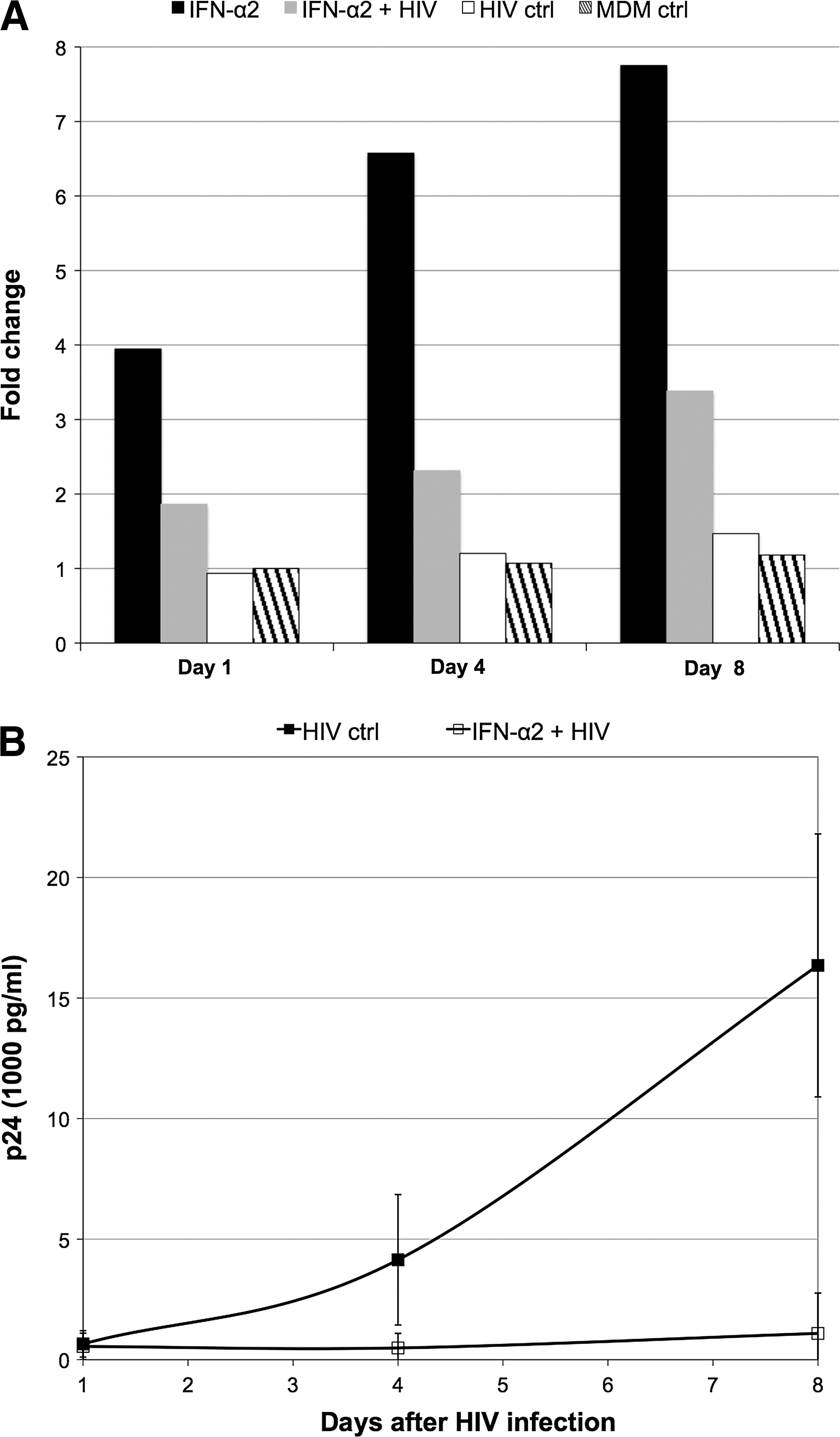
Real-time quantitative polymerase chain reaction (RT-qPCR) validation of OAS1 downregulation by HIV. OAS1 gene expression was assessed by RT-qPCR at all time points (Day 1, 4, and 8) across the experimental conditions described
These studies demonstrated that a 12-h treatment of MDMs with IFN-α2 was the minimal time required to induce maximal protection from HIV replication and that IFN-α2, -α7, -β, and -ω were more potent inhibitors than IFN-α1 and -τ4. In support of this ranking of the inhibitory effects of different species of IFN, Sperber and others (1992) calculated the IFN concentration necessary for 50% protection of MT-2 cells from HIV-induced cytopathic effect (EC50) and demonstrated that IFN-α2 and -α7 were more protective than IFN-α1. Of note, the weaker antiviral nature of IFN-α1 coupled with its low affinity for the human IFN receptor (Uze and others 1985) and ability to bind the type I IFN receptor homolog (i.e., the soluble B18R protein) expressed by vaccinia virus (Liptakova and others 1997), led Mogensen and others (1999) to postulate that IFN-α1 is expressed to negate the effects of such virus-encoded type I IFN receptor decoy molecules. This would facilitate the escape from virus inhibition of other Type I IFNs with greater antiviral activity, which could then signal through the cellular receptor to induce ISGs contributing to innate immunity. Several other studies have simultaneously examined the inhibitory effects of IFN-α2 and -β on HIV replication in primary macrophages (Kornbluth and others 1989; Meylan and others 1993; Cheney and McKnight 2010). Two out of three of these studies (Kornbluth and others 1989; Meylan and others 1993) validate our findings in suggesting that there is little difference between the inhibitory effects of IFN-α2 compared to IFN-β at the concentrations analyzed in our study (1,000 IU). In contrast, Cheney and McKnight (2010) suggested that IFN-α2 was a stronger inhibitor than IFN-β. However, a direct comparison between our studies is difficult because of several differences in the study design of Cheney and McKnight (2010): (1) lower concentrations of IFN, (2) longer periods of IFN-treatment and HIV infection, and (3) assessment of viral replication using a focus forming unit assay. Finally, it has been demonstrated that IFN-ω was capable of inhibiting laboratory and primary isolates of HIV to a similar extent as IFN-α2, although this analysis was performed in PBMCs (Kunzi and Pitha 1996). In sum, these studies lend confidence to our ranking of the inhibitory effect of different species of Type I IFN (Fig. 1) despite the fact that this analysis was performed using MDMs isolated from a single donor.
The major finding of the current study was that the mere presence of HIV in culture was sufficient to downregulate ISGs in MDMs resistant to virus replication through previous exposure to IFN. Since IFN treatment is thought to inhibit HIV replication before proviral integration (Kornbluth and others 1990; Shirazi and Pitha 1992; Meylan and others 1993; Cheney and McKnight 2010), it is plausible that early events in the viral lifecycle (i.e., signaling from the cell surface) may be responsible for the downregulation of ISGs. This has broad implications for HIV infection in vivo, where the virus may have evolved mechanisms to downregulate innate immune responses in target cells already exposed to the Type I IFNs previously released in massive amounts by plasmacytoid dendritic cells following virus detection (Fonteneau and others 2004; Liu 2005). Of particular note, ISG15 and OAS1 are ISGs with known anti-HIV activity (Kunzi and others 1995; Stark and others 1998) and were found to be downregulated by HIV following induction with IFN in this study. Furthermore, OAS (1, 2, and 3) levels appear to be inversely correlated with viral load in HIV-infected individuals treated with IFN-α2a (Asmuth and others 2010). Finally, it was recently shown that short hairpin RNA knockdown of IFI44 following induction by IFN-α2 in SupT1 cells alleviates inhibition of HIV replication (Lu and others 2011).
Determining the selectivity of ISG downregulation by HIV awaits further experimentation with biological replication using MDMs isolated from multiple donors, but probably reflects a general dampening of the IFN-mediated innate immune response or possibly a signaling arm of this response. In summary, the current working hypothesis is that when IFN triggers an antiviral response in macrophages before HIV infection, then the cells are protected from viral replication, but if HIV arrives first, then viral replication rapidly outpaces the innate immune response triggered by IFN (Woelk and others 2004). The avoidance mechanisms that HIV employs while outpacing the IFN response during the natural course of infection are still active against a macrophage cell that has been pretreated with IFN, but appear less effectual and not sufficient enough to facilitate HIV replication.
Footnotes
Acknowledgments
This work was supported financially by an R21 grant from the NIH (AI065242), the Catholic Medical Center Research Foundation, and the Korean Society for AIDS. Experimental assays were performed with assistance from staff at the Genomics Core at the Center for AIDS Research at the University of California San Diego (A136214). J.C. acknowledges the Canada Research Chair in Medical Genomics. This material is based upon work supported, in part, by the Department of Veterans Affairs (VA), Veterans Health Administration, Office of Research and Development, but the views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government. Gratitude is extended to Robert Schooley and David Wyles, who provided advice during the preparation of this manuscript and to Hirotaka Kuwata and Suzanne Gartner who provided advice with respect to experimental assays.
Author Disclosure Statement
None of the authors have any commercial associations that represent a conflict of interest in connection with this body of work. It should be noted that Jean Lozach is currently employed by Illumina, Inc., which is the company that manufactured the microarray platform used in this study. However, this does not represent a conflict of interest since this platform was selected before his inclusion in the study.