
Abstract
Serum response factor (SRF) is required for diverse aspects of development and homeostasis, but potential roles in the regulation of inflammation and immunity have not been systematically investigated. Here, we demonstrate that SRF is unexpectedly required for optimal responses of elicited peritoneal macrophages to type I interferons. Knockdown of SRF expression in these cells impairs induction of numerous interferon (IFN)-stimulated genes (ISGs) in response to zymosan, LPS, and poly I:C. This effect is primarily due to a defect in the ability of induced type I interferons to mediate secondary activation of ISGs. SRF does not appear to be required for expression of established components of the type I interferon signaling pathway, with IFN-β-dependent phosphorylation of STAT1 and STAT2 normally occurring in SRF-depleted macrophages. Collectively, these findings suggest that SRF can indirectly modulate type I interferon-signaling, without interfering with the classic JAK/STAT/ISGF3 pathway.
Introduction
R
Upon ligand binding, TLRs regulate inflammatory responses through activation of downstream signaling cascades to activate numerous downstream transcription factors, including members of the NFκB, AP1, and interferon-regulatory factor (IRF) transcription factor families (Goodridge and Harnett 2005). Upon receptor activation, NFκB, AP1, and IRF proteins are capable of inducing the expression of hundreds of pro-inflammatory genes that comprise the immediate phase of TLR signaling (Li and Verma 2002; Honda and Taniguchi 2006). Products of the primary response genes are involved in initiating secondary responses (Panne and others 2004). TLR induced production and secretion of soluble interferon–β (IFN-β) binds to the interferon α/β receptor (IFNAR) and activates the JAK-STAT pathway, which results in the phosphorylation and translocation of interferon-stimulated gene factor 3 (ISGF3) and induction of interferon-stimulated genes (ISGs) that are important for antiviral host defense (Lehtonen and others 1997; Gale and Foy 2005).
Serum response factor (SRF) is a member of the MADS (
Materials and Methods
Reagents and plasmids
Zymosan A, LPS, polyI:C, IFN-γ, Actinomycin D (Act-D), and cycloheximide (CHX) were obtained from Sigma. Thioglycollate was from BD Biosciences. IFN-α and IFN-β were obtained from PBL InterferonSource.
Expression array profiling
Total RNA (isolated by RNeasy kit; Qiagen) was prepared from untreated or zymosan treated (1 mg/mL, 1 or 6 h) thioglycollate-elicited macrophages. About 0.5 μg of purified RNA per sample was labeled using the LRILAK PLUS, 2 color Low RNA Input Linear Amplification kit and hybridized to an Agilent Whole Mouse Genome Microarray 4×44K 60 mer slides according to the manufacturer's instructions. Slides were scanned using the Agilent GZ505B Scanner and analyzed using Gene Spring Software (Agilent) or DAVID (
Cell culture and transient transfection
Thioglycollate-elicited, peritoneal macrophages were prepared as previously described (Ghisletti and others 2007) from 6–8 week old, male, C57BL/6 mice (Harlan). For RNAi experiments in primary macrophages, 0.75 million cells were transfected with control or SMARTpool siRNAs (100 nM; Dharmacon) directed against Srf mRNA using the Deliver X transfection reagent (Panomics) according to the manufacturer's instructions. Cells were used for experiments 48 h post-transfection and target gene knockdown was validated by Q-PCR.
RNA isolation and Q-PCR
Total RNA (isolated by RNeasy kit; Qiagen) was prepared from primary macrophages. 1 μg of total RNA was used for cDNA synthesis according to the manufacturer's instructions (Superscript III; Invitrogen), and 1 μL of cDNA was used for Q-PCR analysis using SYBRGreenER master mix (Invitrogen) and gene-specific primers. Primer sequences are available upon request. Q-PCR analysis was performed on an Applied Biosystems 7300 Real-Time PCR system. Values were normalized to Gapdh expression (Livak and Schmittgen 2001). Briefly, for each sample, the target gene and the housekeeping gene Gapdh content were both examined by the Real-time PCR system and the CT values were recorded. The relative amount of the target gene was represented by the value normalized to Gapdh (ΔCT=CT(Target gene)–CT(Gapdh), Expression level=2–ΔCT).
Western blotting
For the whole cell lysate analysis, 2–3×106 primary macrophages were lysed with RIPA buffer (40 mM Tris, pH7.4, 150 mM NaCl, 1% Triton X-100, 0.2% SDS, 0.5% Na-deoxycholate, 1× complete protease inhibitors (Roche), 1 mM PMSF, 5 μM E64, and 5 μM MG132). Nuclear and cytoplasmic extracts were prepared from thioglycollate-elicited macrophages using a commercial kit (NE-PER; Pierce). Western blotting was performed as described in detail by Impey and others (2002). Briefly, extracts were resolved by SDS–PAGE (4%–12% NuPAGE; Invitrogen), transferred to PVDF (Immobilon-P; Millipore) and immunoblotted using anti-STAT1 (Santa Cruz), anti-STAT2 (Santa Cruz), anti-p-STAT1 (Santa Cruz), anti-p-STAT2 (Upstate), anti-GAPDH (Abcam), and anti-lamin (Cell Signaling) antibodies. The appropriate alkaline phosphatase-conjugated secondary antibodies (Jackson) were then used and developed with the CDP-Star chemiluminescent reagent (Applied Biosystems) according to the manufacturer's instructions.
De novo motif analysis
Enriched sequence motifs in promoters of differentially expressed genes were identified as previously described (Heintzman and others 2009; Heinz and others 2010; Sullivan and others 2010). Briefly, peak sequences (±200 bp from the center of the peak) were compared to 50,000 random, genomic sequences that were generated to match the size and GC-content of the peak sequences (to remove the sequence bias introduced by CpG islands). Motifs of 8, 10, and 12 base pairs were identified by screening all oligos for enrichment in the target set compared to the background set, with an allowance of 2 bp mismatches to increase the sensitivity of the method. The top 200 oligonucleotides of each length with the lowest p-values were then converted into probability matrices and heuristically optimized to maximize hypergeometric enrichment of each motif in the given data set. As optimized motifs were found they were removed from the data set to facilitate the identification of additional motifs. Sequence logos were generated using WebLOGO (
Results
SRF is globally required for ISG expression in macrophages
To identify transcription factors not previously recognized to contribute to innate immune responses, we performed de novo motif analysis of promoters of genes induced in macrophages by zymosan, a ligand for the dectin-1 receptor and TLR2/6 (Karumuthil-Melethil and others 2008). Inducible promoters were identified using Agilent Mouse Whole Genome Microarrays, using a confident, nonredundant subset of 20,721 probes that could be linked to specific transcriptional start sites (TSS). A total of 472 genes were identified as induced after 1 h of zymosan treatment (Supplementary Table S1-Part I; Supplementary Data are available online at

Genome wide analysis of the role of serum response factor (SRF) in transcription regulation in macrophages.
Based on this observation, we evaluated the impact of knocking down Srf mRNA levels on zymosan responses of elicited mouse peritoneal macrophages. Transfection with SRF-specific siRNAs reduced Srf mRNA levels to approximately 20% of the levels present in macrophages transfected with control siRNAs (Fig. 1B). Control and SRF siRNA-transfected cells were then treated with zymosan for 0, 1, or 6 h and total mRNA was subjected to transcriptome analysis using Agilent microarrays. To identify SRF-dependent genes, we filtered the set of transcripts that were reduced by more than 33% with SRF-specific siRNAs. This resulted in the identification of 263, 370, and 436 genes that exhibited impaired induction upon SRF knockdown at 0, 1 and 6 h of zymosan treatment, respectively. Since we focused our studies on the role of SRF in inflammatory responses, we defined SRF-dependent inflammatory response genes as those transcripts that were induced more than 3-fold after 6 h of zymosan treatment and reduced by more than 33% in macrophages transfected with SRF-specific siRNAs at either 1 or 6 h of zymosan treatment. Filtering the data using these criteria resulted in the identification of 49 genes that exhibited impaired induction upon SRF knockdown at 1 h and an overlapping but distinct set of 64 genes exhibiting impaired knockdown at 6 h (Fig. 1C). Both sets of genes were significantly enriched for functional annotations linked to immune responses based on Gene Ontology analysis using the Gene Spring software (Agilent) (Fig. 1D).
We next applied de novo computational motif discovery methods to search for enriched sequence motifs in the promoters (−500 bp to +500 bp from the TSS) of the SRF-dependent inflammatory response genes identified following zymosan treatment, as described above. Surprisingly, the sequence GAAACTGAAACC, but not a CArG-box, was identified as the most significantly enriched motif in genes exhibiting reduced induction at 6 h (Fig. 1E and Supplementary Table S2), but not at 1 h (data not shown). This motif is nearly identical to the consensus interferon-stimulated response element (ISRE) sequence (ggGAAAGTGAAACCa) (Levy and others 1988). The ISRE is recognized by IRFs and the type I IFN-inducible ISGF3 complex, which is formed by STAT1, STAT2, and IRF9 (Wesoly and others 2007). The identification of the ISRE element is unexpected because SRF has not been reported to biochemically or functionally interact with IRFs or any member of the ISGF3 complex.
SRF regulates the inflammatory response downstream of IFN-β signaling
Consistent with the identification of an ISRE sequence, most of the SRF-dependent, zymosan-induced genes identified by the microarray have previously been identified as ISGs. As the primary effectors of the IFN response, ISGs are indispensable for the antiviral response following viral infection (Takaoka and Yanai 2006). To confirm the time course and SRF dependence of known ISGs, we performed optimized Q-PCR assays to test ISG expression in siRNA treated samples after 0, 1, 3, and 6 h of zymosan stimulation. Knockdown of SRF significantly inhibited the induction of ISGs such as Ifit2, Ifit3, Isg15, Ifit1, Irf7, Isg20, and Oas2 after 6 h of zymosan treatment, but not after 1 or 3 h (Fig. 2A and Supplementary Fig. S1). This result confirms the observation from the microarray data that SRF is required for the full activation of these ISGs. Dectin-1 receptor and TLR2/6 ligands are not considered to be typical type I interferon response inducers. Consistent with this, interferon beta expression was only weakly induced (<3-fold) 3 or 6 h following stimulation with Zymosan or FSL-1 (data not shown). We therefore examined the effect of SRF on the expression of ISGs after activation by polyI:C (recognized by TLR3) and LPS (recognized by TLR4). These experiments demonstrated that the loss of SRF also impaired ISG induction in response to TLR3 and TLR4 signaling (Fig. 2B, C), indicating that SRF has a general role in regulating the interferon response downstream of TLR activation.
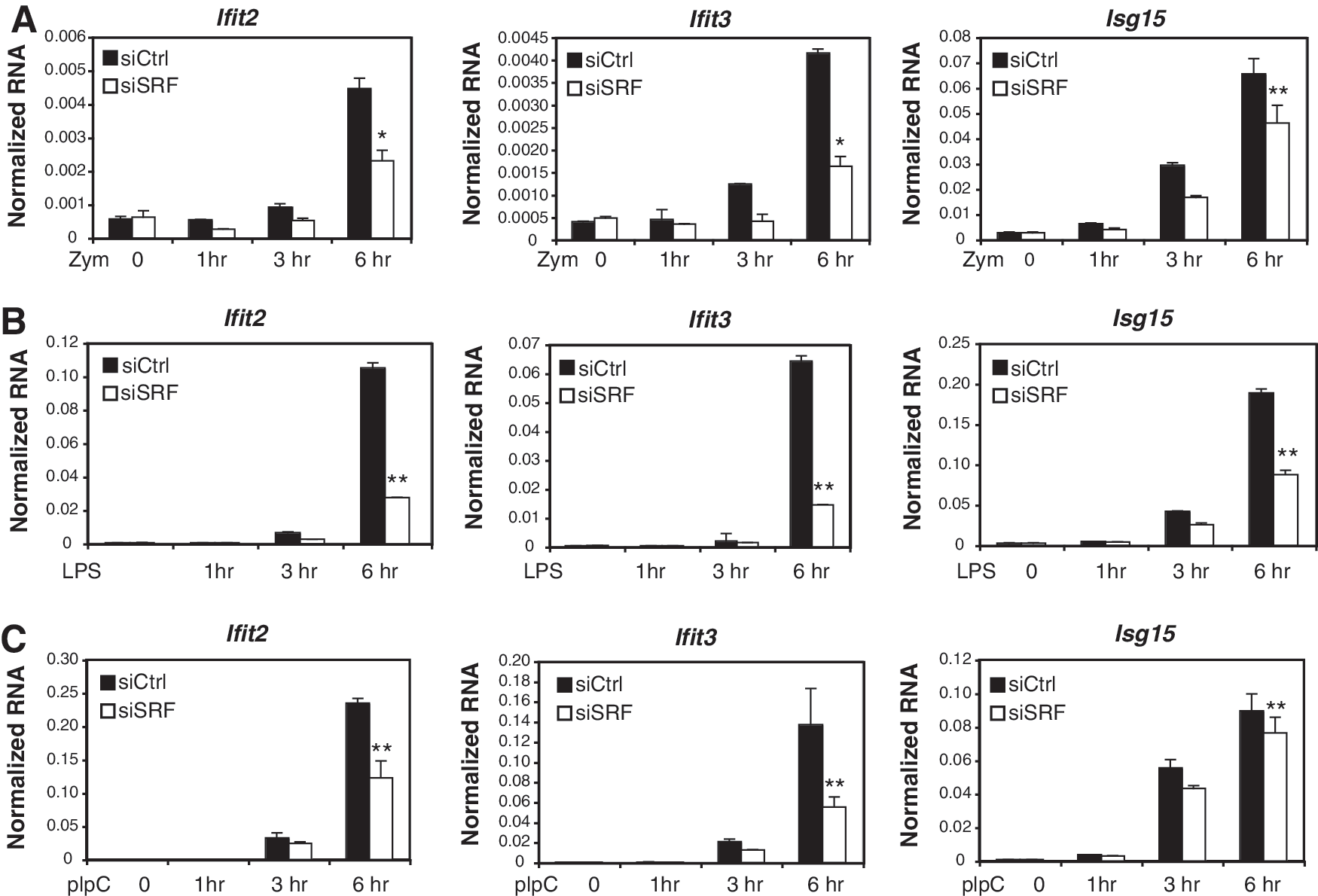
Expression profiles of SRF-dependent interferon-stimulated genes (ISGs).
SRF effect on ISG expression is specific for type I interferon-signaling
To determine whether SRF functions upstream or downstream of interferon receptor signaling, we used IFN-β and IFN-α to directly induce ISG expression in control and SRF knockdown cells. These experiments demonstrated that ISG expression was also impaired following SRF knockdown, suggesting that SRF acts downstream of type I interferon signaling to influence ISG responses (Fig. 3A, B). Interestingly, SRF-dependent ISG expression is specific for type I interferon-signaling because SRF siRNA-transfected cells treated with type II IFN-γ did not exhibit defects in the induction of well-established IFN-γ target genes, including Irf1, Oas2, and Stat1 (Fig. 3C).

SRF regulation of ISGs is specific for type I interferons.
SRF knockdown antagonizes ISG expression independent of new protein synthesis and without affecting mRNA stability
To further examine SRF function in the interferon response, we tested whether the impairment of ISG expression in response to SRF knockdown was dependent on new protein synthesis. We hypothesized that SRF loss may affect intact ISG expression through either a defect of a secondary activator that is induced after IFN-β stimulation or through the increased expression of negative regulators of the interferon pathway such as SIGIRR (single immunoglobulin IL-1R-related molecule) or SOCS (Suppressor of cytokine signal) proteins (Coccia and others 2006). To exclude the possibility of the upregulation of a negative regulator following SRF knockdown, we pretreated control and SRF knockdown primary macrophages with cycloheximide to block new protein synthesis and then stimulated the cells with IFN-β. For the IFN-β-treated cells, SRF was required for an intact interferon response of Ifit2 both in the presence and absence of cycloheximide pretreatment indicating that SRF affected ISG transcription independent of new protein synthesis (Fig. 4A). The same result was seen for genes Ifit1 and Oas2 (data not shown). As a control for the effectiveness of the cycloheximide blockade, we also stimulated control and SRF knockdown primary macrophages with polyI:C after cycloheximide pretreatment. In this case, the induction of Ifit2 expression was impaired by cycloheximide pretreatment. This result is consistent with a block in the production of the type I interferons required for the secondary expression of ISGs and confirmed that new protein synthesis was indeed blocked in our experiment (Fig. 4B).
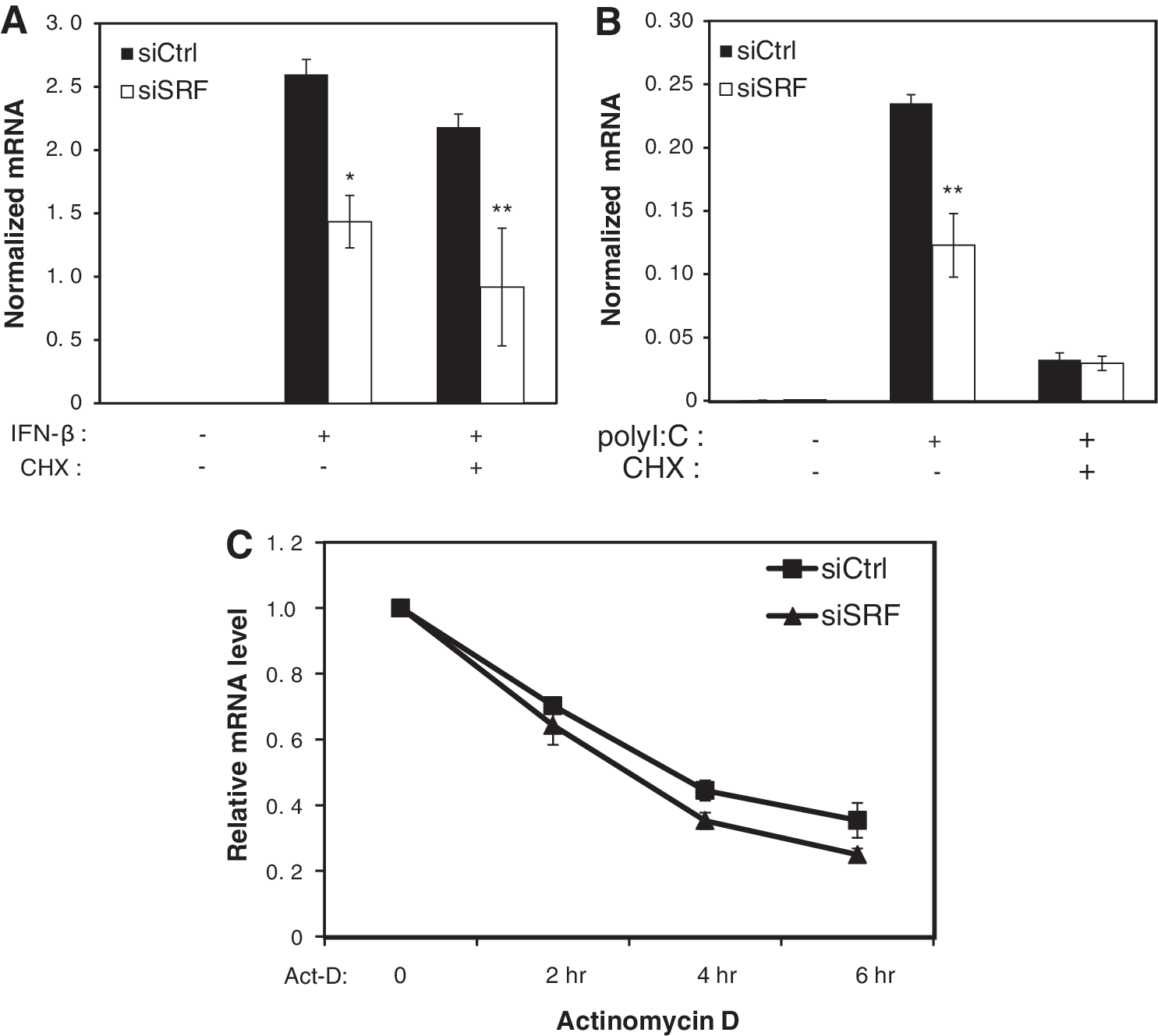
SRF affects ISGs independent of protein synthesis and mRNA stability.
Experiments were also performed to determine whether the defects in the accumulation of ISG mRNA after SRF loss were due to decreased transcription of the mRNA or decreased mRNA stability. To distinguish these possibilities, control and SRF-specific siRNA knockdown macrophages were first treated with IFN-β (1000 U/mL) for 6 h to induce an initial wave of transcription and then treated with the transcription inhibitor, Actinomycin D (Act-D), to block transcription. RNA was harvested at 0, 2, 4, and 6 h after Act-D treatment, respectively, and Ifit3 expression was analyzed. Our results indicated that there were no significant differences in the Ifit3 mRNA levels in the control or SRF-specific siRNA-transfected samples following Act-D treatment relative to the induced Ifit3 expression levels (Fig. 4C). These results suggest that SRF knockdown does not cause decreases in ISG expression through increased mRNA turnover.
SRF does not affect the IFN-JAK-STAT-ISGF3 pathway
One possible mechanistic explanation for the observed defect in ISG activation after SRF loss could be the negative regulation of the JAK-STAT-ISGF3 signaling pathway. The effect of SRF expression on type I interferon signal transduction was tested by examining the expression, phosphorylation, and nuclear accumulation of STAT1 and STAT2 after IFN-β stimulation. Western blots performed on macrophage whole cell extracts showed that protein levels of STAT1 and STAT2 are somehow increased with SRF knockdown in untreated conditions and are not changed with or without SRF knockdown after IFN-β stimulation. Moreover, immunoblotting with anti-phospho-STAT1/2 antibodies indicated that the IFN-β-dependent activation of STAT1 and STAT2 was also not inhibited by SRF loss (Fig. 5A). The subcellular distributions of STAT1 and STAT2, and the phosphorylated STAT proteins, were also examined and showed no dependence on the presence of SRF, both at an early time point (30 min, Fig. 5B) and a late time point (18 h, data not shown). These results indicate that IFN-β-responsive signal transduction from the cytoplasm to the nucleus remains intact after SRF loss, suggesting that SRF is required in another intra-nuclear step in ISG induction.

SRF affects ISGs without affecting the expression, phosphorylation, and nuclear translocation of ISGF3 signaling molecules.
To identify whether SRF directly regulates ISG expression, we checked SRF binding sites on a genome-wide scale using chromatin immunoprecipitation coupled to deep sequencing (ChIP-Seq) in primary macrophages. Our ChIP-Seq analysis of SRF resulted in the identification of more than 1,000 binding sites (Sullivan and others 2010), including binding sites in the well-known SRF target genes cfos (Treisman 1986) and vinculin (Vcl) (Cen and others 2003). However, none of the SRF-dependent ISG promoters showed occupancy by SRF (data not shown), consistent with an indirect effect of SRF on the transcription of ISGs.
Discussion
While essential roles of SRF in embryogenesis, neuronal development, smooth muscle differentiation, and cardiac cell differentiation have been extensively documented (Miano 2003), much less is known concerning the functions of SRF in the immune system. Studies in lymphocytes have demonstrated that T cell-specific deletion of Srf results in a severe block in thymocyte development at the transition from CD4/CD8 double to single positive stage, while B cell-specific Srf deletion leads to a complete loss of marginal zone B cells and a marked reduction of the CD5+B cell subset (Fleige and others 2007). These results indicate essential and distinct functions of SRF in the differentiation of T and B cells. Additional evidence relating SRF to immune system function was provided by a study demonstrating that LPS activation of tissue factor and TNF-α expression in monocytic cells involved SRF-dependent induction of egr-1 (Guha and others 2001).
Here, we provide evidence that SRF is required for optimal transcriptional responses of primary macrophages to type I interferons. Transcriptional profiling of zymosan-treated macrophages led to the identification of a CArG-box motif as being significantly enriched in upregulated promoters. Transcriptome analysis of SRF-deficient cells also indicated that SRF is required for the induction of both early (1 h) and late (6 h) subsets of zymosan-inducible genes. Unexpectedly, de novo motif analysis of late inducible gene promoters dependent on SRF for induction revealed an ISRE sequence as the most highly enriched transcription factor binding site, suggesting a role of SRF in regulating interferon-dependent gene expression. These results were confirmed and extended by the demonstration of a requirement for SRF for full induction of numerous ISGs in response to zymosan, polyI:C, and LPS. Based on our findings, it is tempting to speculate that SRF function is important for immune responses against viral infection, since the virus-triggered IFN-β pathway is critical for host survival.
Molecular analysis indicates that the impairment of ISG induction in SRF-deficient macrophages in response to TLR activation primarily results from a defect in the ability of induced type I interferons to mediate secondary gene expression. Intriguingly, this effect is limited to the type I interferon pathway, as type II interferon-induced target gene activation was not affected by SRF knockdown. The underlying mechanism responsible for this defect remains to be established. Our studies showed that phosphorylation and nuclear translocation of STAT1/2 in response to IFN-β normally occurs in SRF-deficient macrophages. We also excluded the possibility that loss of SRF could affect the persistence of ISGF3 in the nucleus, because the amount of phosphorylated STAT1/2 in the nucleus was not changed in the absence of SRF up to 18 h following IFN-β treatment. Even though SRF ChIP-Seq did not reveal direct SRF binding to these ISRE-containing genes, it is possible that SRF could act as a non-DNA binding coactivator to promote transcription complex formation with STATs. Alternatively, SRF may be required for proper expression of a co-activator that is required for persistent gene activation. Further studies are needed to investigate the complete mechanism of how SRF is involved in the regulation of ISG expression.
We previously demonstrated that SRF primarily regulates cytoskeleton-dependent functions in macrophages under basal conditions (Sullivan and others 2010). Here, our studies suggest that SRF regulates an inflammatory gene program, in addition to the immediate early genes and cytoskeletal genes that have already been described. These findings suggest that SRF is involved in the establishment of an antiviral state in macrophages, and thus is an important factor for regulating innate immunity.
Footnotes
Acknowledgments
We are grateful to Dr. Christopher Benner and Dr. Michael David for helpful discussion and suggestions. We thank the UCSD Biogem Core facility for labeling and hybridization of the expression microarray samples. We also thank Lynn Bautista for her assistance with preparation of the figures. This work was supported by NIH grants CA52599 and DK063491.
Author Disclosure Statement
No competing financial interests exist.