
Abstract
Cystic fibrosis (CF) is due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which cause a massively proinflammatory phenotype in the CF airway. The chemical basis of the inflammation is hyperproduction of interleukin-8 (IL-8) by CF airway epithelial cells, based on both an intrinsic mutation-dependent mechanism and by infection. In infection-free, cultured CF lung epithelial cells, high levels of the microRNA (miR), miR-155, is responsible for hyperexpression of IL-8. However, whether infection-induced IL-8 expression in CF cells is also mediated by miR-155 is not known. We have hypothesized that miR-155 might be a general mediator of enhanced IL-8 expression in CF cells, either in response to other cytokine/chemokine mediators of inflammation, or after exposure to infectious agents. Here we find that a reduction in miR-155 accompanies suppression of IL-8 by either the anti-inflammatory cytokine IL-10 or by inhibition of ambient IL-1β with a neutralizing antibody. However, attempts to elevate IL-8 levels with either intact bacteria [viz. a mucoid strain of Pseudomonas aeruginosa (PA)], or lipopolysaccharide were unable to elevate miR-155 above its intrinsically high level in the absence of these agents. Instead, in response to PA infection, the CF cells modestly suppress the expression of miR-155, and express a novel set of miRs, including miR-215. We find that ex vivo CF lung epithelial cells also express high levels of both miR-155 and miR-215. The predicted module of infection-induced mRNA targets focuses on activation of the NFκB-signaling pathway, and on the proapoptotic p53-signaling pathway. We interpret these data to suggest that that CF lung epithelial cells respond to PA or bacterial cell products with a novel miR program that may carry with it serious challenges to survival.
Introduction
C
However, the airway of the patient with CF is chronically infected, most often with mucoid strains of Pseudomonas aeruginosa (PA). This chronic infectious state therefore adds to the already high IL-8 expression level by added signaling through the toll-like receptor (TLR)- and NFκB-signaling pathways. Therefore, a combination of defective intrinsic regulation of IL-8 expression, coupled with infection-mediated increase in IL-8 expression, may result in a catastrophic proinflammatory quagmire of chemokine activities, bacteria and bacterial products, neutrophils, and mucins in the CF airway. However, it remains unknown whether a common intrinsic defective mechanism is responsible for both the intrinsic defect in IL-8 expression, as well as the defective response to the extracellular presence of bacteria, in the CF airway.
The expression of inflammatory genes, including the proinflammatory chemokine IL-8, is known to be regulated by diverse processes. We have previously demonstrated that CF lung epithelial cells in culture not only secrete large amounts of IL-8 protein, but also have high levels of very stable IL-8 mRNA (Balakathiresan and others 2009). Recent studies have also shown that IL-8 expression in CF cells is regulated by the microRNA, miR-126 (Oglesby and others 2010). More recently, we have specifically shown that miR-155 is directly responsible for stabilizing IL-8 mRNA and upregulating of IL-8 protein expression in CF lungs (Bhattacharyya and others 2011). Elevated levels of miR-155 are also found in primary cultures of airway epithelial cells from patients with CF, as well as in neutrophils from peripheral blood of patients with CF (Bhattacharyya and others 2011). However, whether this mechanism is also responsible for massively elevated levels of IL-8 in response to bacteria has not yet been determined.
We have therefore hypothesized that miR-155 might be a general mediator of enhanced IL-8 expression in CF cells, including in response to other cytokine/chemokine mediators of inflammation, or after exposure to infectious agents. To test this hypothesis, we have challenged CF lung epithelial cells with conditions that either raise or lower IL-8, and have measured changes in miR-155. Here, we report as anticipated that agents such as the anti-inflammatory cytokine IL-10, or neutralizing antibodies against IL-1β, lower both IL-8 and miR-155. However, while PA raises IL-8 levels, we find that it fails to further elevate miR-155. Instead, PA modestly suppresses miR-155 and recruits an entirely unique set of miRs. This unique set of miRs includes elevated miR-215, whose focus with other PA-dependent miR changes is to drive the CF cell into a p53-centric survival challenge. These data suggest that CF is associated with both an intrinsic proinflammatory defect, and an infection-elicited defect, and is therefore of importance in resolving this ongoing controversy.
Materials and Methods
Reagents
LHC-8 media, Trypsin-EDTA (0.05%), and Lipofectamine transfection reagent were purchased from Invitrogen. Actinomycin D was purchased from Sigma Chemical Co. RNA aqueous and miRVana kit for isolation of RNA from CF cells was obtained from Life Technology. Anti-IL-1β antibody was purchased from R&D Systems. The human IL-10 was purchased from Cell Signaling Technology, Inc.
Cell culture and transfection
IB3-1 CF lung epithelial cells were maintained in an LHC-8 serum-free medium in humidified 5% CO2 as previously described (Eidelman and others 2001). Transfections were performed using the Lipofectamine Transfection Reagent (Invitrogen). The PA infection was performed according to published protocol (Chattoraj and others 2011). Briefly, 1.5×106 cells were treated with high (HA)- or low (LA)-algenate form of mucoid PA (MPA) for 24 h. RNA was isolated and analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). The dosage of bacterial treatment is nontoxic to the CF cells and was selected based on the published protocol of Chattaraj and others (2011).
Measurement of mRNA and miR by qRT-PCR
RNA was isolated from the CF cells, and mRNA expression levels were analyzed with qRT-PCR as described earlier (Balakathiresan and others 2009). The primary bronchial epithelial cells were obtained from lung brush biopsies of patients with CF under an USUHS Institutional Review Board-approved protocol as described earlier (Balakathiresan and others 2009). The cells were stored in RNA later or Trizol, and total RNA was isolated using the miRVana kit. The miR expression profile was analyzed by TaqMan Low-Density Arrays (ABI) as described in Bhattacharyya and others (2011). Briefly, 350 ng of RNA was used for the profiling. Each of the multiplex reverse transcription performed with the TaqMan miRNA Reverse Transcription Kit (ABI) was diluted and mixed with TaqMan Gene expression Master Mix (2×). One hundred microliters of the RT reaction-specific PCR mix was loaded into the corresponding fill ports of the TaqMan Low-Density Human MicroRNA Panel v2.0 (Early Access). The individual miR-specific TaqMan assays were performed with 20 ng of RNA with TaqMan probes specific for mature miRs (ABI).
Statistical analysis
Statistical analysis was performed using Excel. Significance values (P≤0.05) were determined by Student's t-test for the 3-h time points. Error bars on graphs represent SEM.
Results
IL-10 treatment of CF cells suppresses IL-8 and miR-155 expression
CF epithelial cells are also functionally deficient in IL-10, an important anti-inflammatory, immune-modulatory cytokine (Virella-Lowell and others 2004). To test if IL-10 treatment could suppress IL-8 in CF lung epithelial cells, we treated IB3-1 CF cells with IL-10 (10 ng/mL) for various intervals of time. We subsequently measured IL-8 mRNA levels. As depicted in Fig. 1a, we find that a 2-h incubation of the CF cells with 10 ng/mL IL-10 promotes the most significant and efficient reduction in IL-8 mRNA. Figure 1b shows that IL-10 treatment also suppresses miR-155 by ∼20% (P<0.05). There is precedence for this kind of observation, since IL-10 has been shown to suppress miR-155 expression in mouse macrophages (McCoy and others 2010).

Effect of interleukin (IL)-10 on IL-8 expression in cystic fibrosis (CF) cells.
We have previously shown that the miR-155 mechanism for IL-8 activation depends on suppression of the inositol 5-phosphatase SHIP1, resulting in activation of the PI3K/Akt/MAPK signaling pathway (Bhattacharyya and others 2011). Activation of MAPK by this mechanism results in stabilization of the IL-8 mRNA. Consistently, Fig. 1c shows that reduction of miR-155 through treatment with IL-10 also results in enhanced degradation of IL-8 mRNA. Thus, the miR-155 mechanism contributes to IL-8 suppression by IL-10.
Anti-IL-1β suppresses IL-8 gene expression and miR-155 in CF lung epithelial cells
IL-1β has been shown to induce IL-8 expression in CF cells (Muselet-Charlier and others 2007) and also to promote stabilization of IL-8 mRNA in various cell types (Suswam and others 2005). We therefore examined the role of IL-1β in the hyperexpression of IL-8, as well as in the upregulation of miR-155 by treating CF cells with different doses of neutralizing anti-IL-1β antibody for 24 h. As shown in Fig. 2a, a dose of 50 ng/mL of the antibody (1:10,000-fold dilution) is effective in suppressing IL-8 expression. Treatment with this dose of anti-IL-1β reduces IL-8 mRNA levels by ∼60% and protein levels by ∼75%. Subsequently, we examined the stability of IL-8 mRNA at this dose of antibody treatment. As shown in Fig. 2b, neutralization of IL-1β promotes enhanced degradation of IL-8 mRNA (P<0.05). There is rapid reduction of IL-8 mRNA within 1 h, followed by decay of IL-8 message to ∼65% of its original level. This rate and extent of IL-8 mRNA decay are comparable to that observed in CFTR-repaired IB3-1/S9 lung epithelial cells (Balakathiresan and others 2009). These data have historical precedents in earlier studies of other cell types showing effects of anti-IL-1β on both IL-8 mRNA transcription and mRNA stability (Suswam and others 2005; Muselet-Charlier and others 2007).

Effect of inhibiting IL-1β on IL-8 mRNA stability and IL-8 protein in CF lung epithelial cells.
Based on our earlier findings that IL-8 expression is regulated by miR-155 in CF cells, we further analyzed miR-155 expression levels in the anti-IL-1β-treated IB3-1/CF cells. Figure 2c shows that miR-155 expression is indeed suppressed by neutralizing IL-1β. However, the level of miR-155 suppression is reduced by only ∼20%. Nonetheless, the reduction is significant (P<0.05), and there are precedents in the literature for small changes in miR expression to have significant impact on target genes (Selbach and others 2008). Conclusively, these data strongly support the concept that elevated IL-1β is one of the factors in CF cells, which promotes hyperexpression of IL-8. These data are also consistent with the possibility that miR-155 expression helps to mediate this effect on IL-8 mRNA expression and mRNA stability in CF lung epithelial cells.
PA infection elevates IL-8 expression, and enhances IL-8 mRNA stability, but decreases miR-155
HA strains of MPA are often closely associated with CF lung disease. We therefore examined the effects of both low-alginate (LA) as well as the HA forms of MPA on IL-8 expression in CF lung epithelial cells. As shown in Fig. 3a, the HA form of MPA induced the most significant increase in IL-8 mRNA levels in IB3-1 CF cells. In as much as increased IL-8 expression in the infection-free state is, in part, due to enhanced stabilization of the IL-8 message (Balakathiresan and others 2009), we proceeded to examine whether MPA infection had any effect on IL-8 mRNA stability.
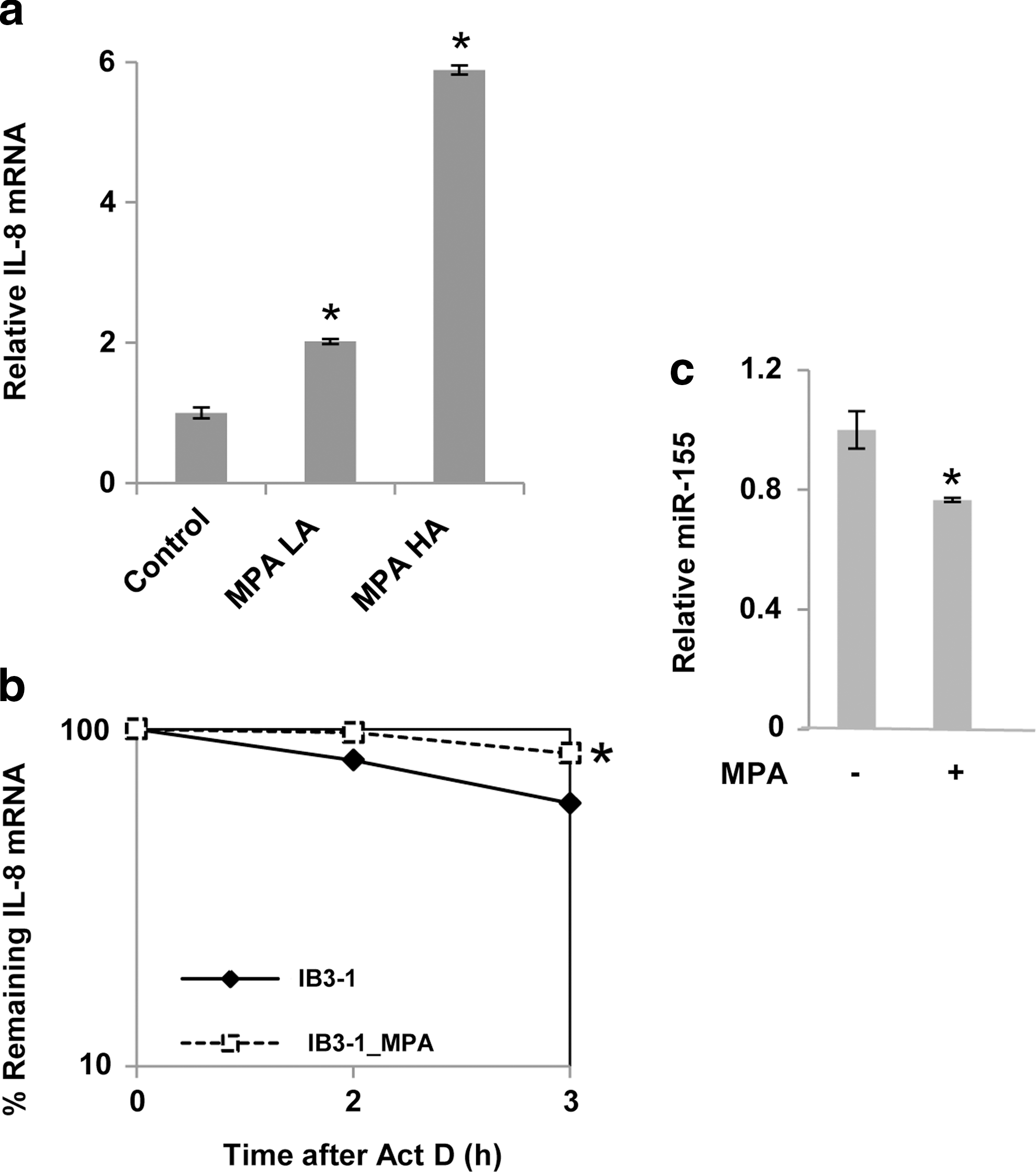
Mucoid Pseudomonas aeruginosa (MPA) induces increased stability of IL-8 mRNA in CF cells.
As shown in Fig. 3b, IL-8 mRNA stability is further enhanced (by ∼3-fold increase in t 1/2) in MPA-treated cells, compared to medium-treated CF cells. The log/linear plot shows that even though IL-8 mRNA stability is the CF cells is quite high to begin with, IL-8 stability can be even further enhanced by the added presence of bacteria. However, as shown in Fig. 3c, we find that increased expression of IL-8 mRNA in the MPA-infected CF cells does not promote further increase in miR-155 expression. Rather, it modestly, but significantly, suppresses miR-155 expression. These data suggest that the miR-155-dependent mechanism for regulation of IL-8 expression in infection-free CF cells might be modified by the concomitant presence of infection.
Lipopolysaccharide further stimulates IL-8 mRNA expression and IL-8 mRNA stability in CF cells without inducing miR-155
Dead bacterial cell walls, represented by purified lipopolysaccharide (LPS), can interact with specific TLRs on the epithelial cell surface, and promote proinflammatory intracellular process that are similar to those evoked by living bacterial cells. We therefore treated CF lung epithelial cells to LPS, and analyzed the IL-8 mRNA response. Figure 4a shows that IL-8 mRNA levels are significantly increased in IB3-1 cells over many hours. However, Fig. 4b shows that no significant increase is observed in the corresponding miR-155 expression levels over the same time period. Finally, Fig. 4c shows that LPS significantly stabilizes IL-8 mRNA compared to the already-stabilized levels observed in medium-treated CF cells. Thus, the miR-155-independent effects of living MPA on IL-8 mRNA levels, IL-8 expression, and IL-8 stabilization can be similarly observed in the presence of a purely chemical stimulus by LPS.
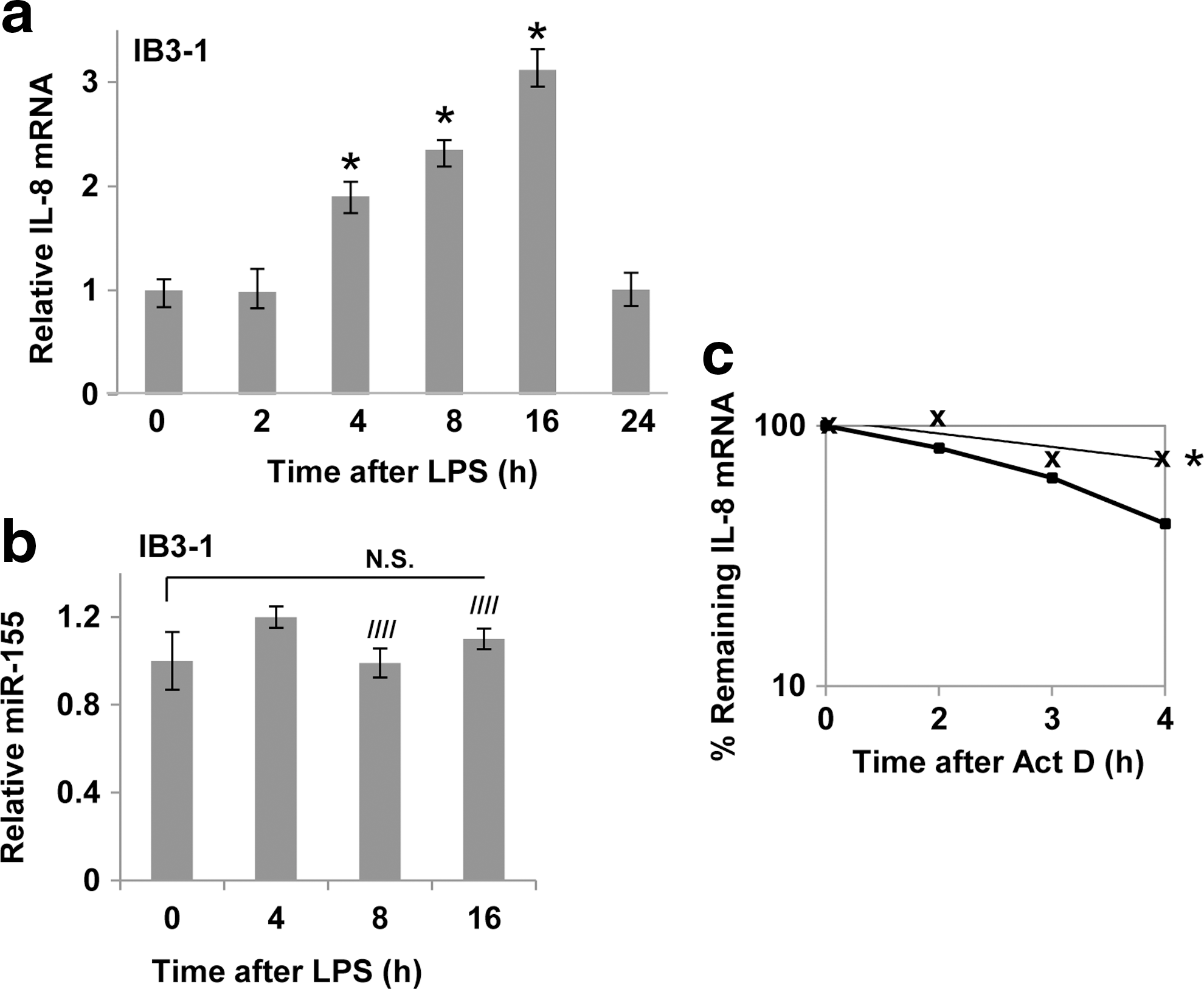
Lipopolysaccharide (LPS) induces increased IL-8 expression in CF cells without affecting miR-155.
Differential expression of miRs in MPA-infected CF lung epithelial cells
Based on the preceding data with intact bacteria and purified LPS, we considered the possibility that the added presence of infection modifies the miR-155 dependent, intrinsic proinflammatory program in CF lung epithelial cells. To test this hypothesis further, we analyzed the miR expression profile in IB3-1 CF treated with MPA, and compared that to medium-treated IB3-1 CF cells. As shown in Table 1, the difference is substantial and mechanistically informative. The data indicate that 75 miRs are significantly (P<0.05) and differentially expressed in CF IB3-1 cells treated with MPA bacteria for 24 h. Of these significantly changed miRs, miR-215 is the only miR whose expression is elevated (∼10-fold), while the other miRs are downregulated compared to uninfected IB3-1 CF cells. According to the prevailing paradigm for understanding miR function, an elevated miR would suppress target mRNAs, while reduced miRs would release target mRNAs from tonic inhibition.
miR-215 is upregulated (
CF, cystic fibrosis; miRNA, microRNA; mucoid Pseudomonas aeruginosa.
To determine the possible relevance of these miR data to clinical CF, we performed the same screen on ex vivo CF lung epithelial cells. As shown in Fig. 5a, we found that miR-215 was also elevated by ∼1,000-fold in CF cells, compared to control cells. In as much as most patients with CF have a history of chronic PA infection, the in vivo miR phenotype, at least with regard to miR-215, appears to parallel, in some fashion, infection-treated IB3-1 cells. miR-215 has been shown to be induced by the tumor suppressor gene p53 (Pichiorri and others 2010). Consistently, we find an ∼2-fold increase in p53 expression in the PA-infected IB3-1 CF cells compared to uninfected control cells and ∼3-fold increase in p53 in CF brush-biopsy cells compared to control normal human bronchial epithelial cells (Fig. 5b).
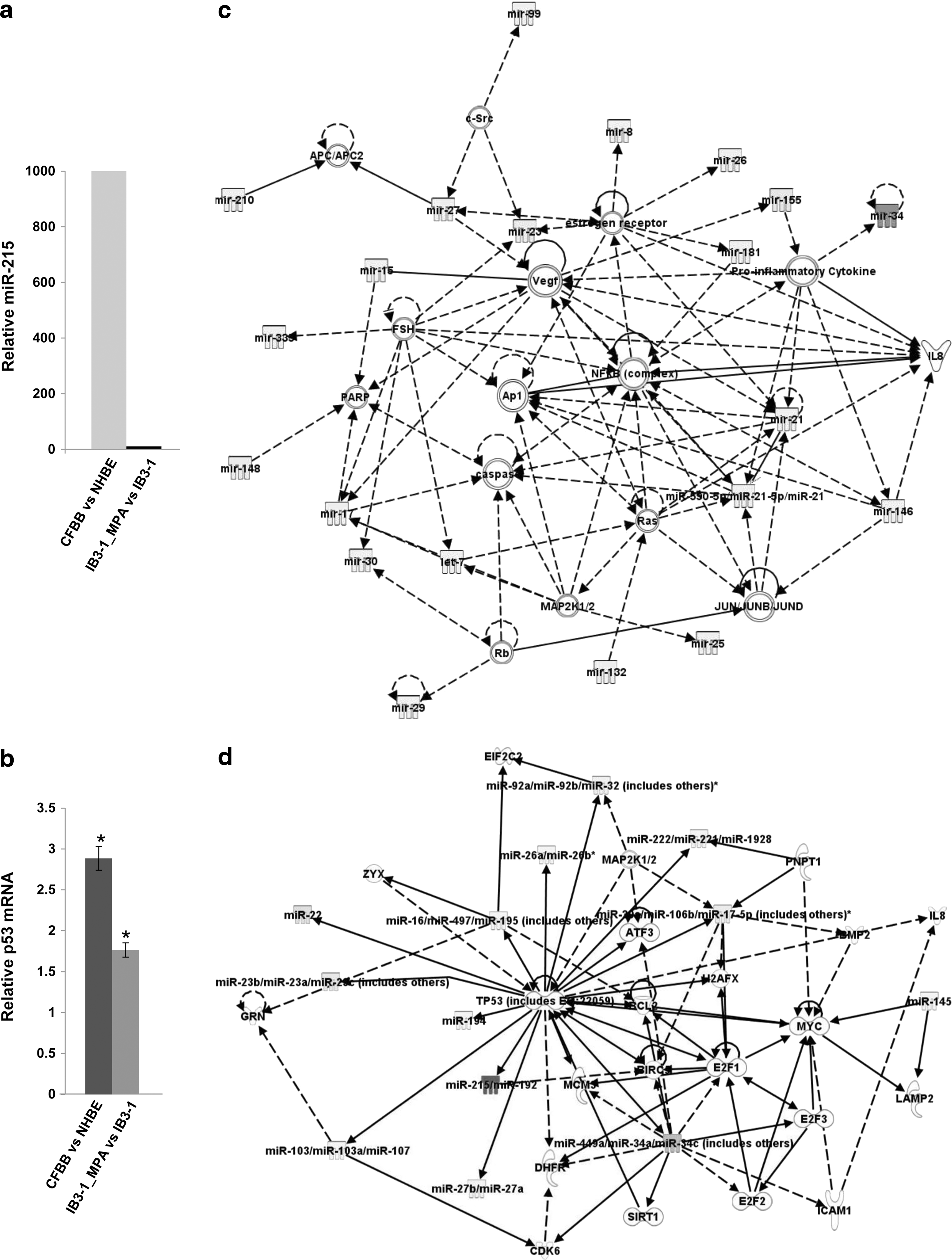
miRNA expression profile in PA-infected CF cells.
To further investigate the mechanistic significance of the entire 88-member miR pattern, including miR-215, we performed a set of network analyses using the ingenuity pathway analysis (IPA) algorithm. As shown in Fig. 5c, IPA identifies a network of miRs that target messenger RNAs for proinflammatory signaling, including MAPKs, NFκB, and IL-8. Since aside from miR-215, all the infection-induced miRs are reduced, the objective trajectory of the predicted, composite signaling ensemble is to drive inflammation. The IPA algorithm also identifies other signaling networks, although at lower levels of significance. In the present case, Fig. 5d shows that the ensemble of infection-mediated miRs forms a significant network that also focuses the tumor suppressor gene p53 (TP53). The p53 network includes 4 miRs (in addition to the 75) that were largely suppressed by PA infection and were therefore detected only in the uninfected IB3-1 CF cells (highlighted in gray, Table 1). Consistently, deletions and mutations in the TP53 gene have also been shown to promote dysregulated inflammatory signaling (Staib and others 2005; Patel and others 2011). In addition, p53 expression has been shown to be affected by certain pathogens, including human papilloma virus (Patel and others 2011). Moreover, p53 has been shown to induce miRs, including miR-215 (Pichiorri and others 2010). Additionally, p53-responsive miRs, including miR-215, have been shown to regulate the cell cycle (Braun and others 2008; Georges and others 2008). Thus, the miR-155-dependent proinflammatory signaling pathway characterizes infection-free CF lung epithelial cells. However, when CF lung epithelial cells encounter infection, the response appears to be supplemented by a different, but still proinflammatory, miR-215-based signaling pathway.
Discussion
In this article, we have tested the hypothesis that miR-155 might be a general mediator of enhanced IL-8 expression in CF lung epithelial cells, either after exposure to cytokine/chemokine mediators of inflammation or in response to infectious agents. We have previously reported that miR-155 stabilizes IL-8 mRNA, thereby enhancing IL-8 protein expression. Consistently, abnormally high levels of miR-155 have been shown in cultured CF lung epithelial IB3-1 cells, primary epithelial explants from CF lung, and acutely isolated, peripheral blood neutrophils from CF plasma (Bhattacharyya and others 2011). The data clearly show here that in the infection-free state, IL-10 mediates reduction in IL-8 mRNA expression, as well as miR-155 levels. The data further show that reduction in IL-1β with a neutralizing antibody against IL-1β has the same concurrent effects on suppression of both IL-8 and miR-155. However, exposure to either intact PA or LPS does raise IL-8 mRNA expression, but does not raise levels of miR-155 above the already high levels. In fact, miR-155 is somewhat suppressed in PA-infected CF cells. Instead, the miR-155 response appears to be supplemented, or replaced, by a different, but still proinflammatory, miR-215-based signaling pathway, which targets both NFκB- and p53-signaling pathways. The increased targeting of NFκB is not so surprising, since the TLRs, which respond to intact bacteria and to LPS, feed downstream into the NFκB pathway through IKK (Berube and others 2010). However, the additional focus on p53 is unexpected, and suggests that the new miR-based signaling pathways drive the CF cell into a p53-centric survival disadvantage when encountering infection. Importantly, the clinical relevance of these new observations is evident from the fact that a significant elevation in miR-215 (∼1,000-fold) is also observed in ex vivo primary lung epithelial cells.
Elevated miR-155 signaling in infection-free CF lung epithelial cells
The possibility of an intrinsic proinflammatory defect in CF lung epithelial cells has been the cause of ongoing controversy in the CF field for many years (Tirouvanziam and others 2000; Eidelman and others 2001; Ribeiro and others 2005; Machen 2006). However, these data provide added support for the concept that the absence of infection, elevated miR-155, as previously reported (Bhattacharyya and others 2011), plays a direct role in the intrinsically elevated state of IL-8 expression in CF lung epithelial cells. The miR-155-encoding gene, BIC, was originally discovered as an oncogene for B cell lymphomas in chicken (Clurman and Hayward 1989), and is also found to be elevated in human B-cell lymphomas (Eis and others 2005). By contrast, in noncancer states, substantial elevation in miR-155 expression can be induced in cells of the innate immune system by many types of proinflammatory stimuli (O'Connell and others 2007). Among others, sensitive proinflammatory systems include TLR3, a receptor for poly(I:C); TLR4, a receptor for LPS; TLR9, a receptor for hypomethylated CpG; TLR2, a receptor for lipoprotein; IFN-β; TNF-α autocrine/paracrine signaling after stimulation by IFN-γ; and by activation of the JNK and NFκB signaling pathways. However, in the case of infection-free cultures of CF lung epithelial cells, the intrinsic elevation of miR-155 would appear to suggest that the system is responding as if bona fide, yet unknown, proinflammatory stress signals were active. However, what that intrinsic signal might be, dependent as it is on the expression of mutant CFTR, is evidently not a response to inadvertent inclusion in the culture of bacteria such as PA, or bacterial products such as LPS. These agents activate yet another, or an additional, set of miRs, including miR-215.
Elevated miR-215 signaling in infected CF lung epithelial cells
miR-215 was originally discovered as a p53-inducible miR, which mediates the p53 effect of cell cycle arrest at both G1 and G2M (Georges and others 2008). From this perspective, miR-215 has been viewed as a tumor suppressor gene in its own right, and responsible for the extensively characterized transcriptional repression functions of p53. However, CF lung epithelial cells are not cancer cells. Consistently, levels of miR-215 in these cells are low. Yet, unexpectedly, in the presence of either HA PA, for 24 h, the levels of miR-215 in the cultured cells become elevated by ∼10-fold. Just as unexpectedly, we find that miR-215 levels in ex vivo CF lung epithelial cells are elevated by ∼1,000-fold over control cells. Prospectively, we had chosen the 24-h exposure of the cultured CF lung epithelial cells to model the chronic infectious state of the CF airway. It is therefore possible that the common elevation of miR-215 in the in vivo and in vitro conditions of CF lung epithelial cells is consistent with expectations. Thus, on the basis of pure discovery science, miR-155 would appear to be a biomarker for the absence of infection in CF cells, while miR-215 appears to be a biomarker for the presence of chronic infection.
However, at the level of mechanism, we may inquire how chronic/long-term infection of CF lung epithelial cells with PA, and/or exposure to LPS, leads to elevation of miR-215, strongly enhanced IL-8 expression, and a highly significant association with p53 signaling among many of the novel miRs coinduced with miR-215. One possibility rests in the fact that oxidative stress is among the principal drivers, including radiation and genotoxic drugs, for p53 expression (Vogelstein and Kinzler 2001; Oren 2003; Jonker and others 2012). Uninfected cultures of CF lung epithelial cells express a chronic phenotype of low-level oxidative stress termed ER stress. This condition is thought to be based on the unfolded protein response associated with the retention of the folding mutant [ΔF508]CFTR in the ER, and elevated levels of calcium (viz, [Cai ++]) in the ER. Under these conditions, we have demonstrated here that miR-155 is elevated, and miR-215 is low. However, as has been previously reported by Ribeiro and others (2005), high levels of calcium are found in the ER of infected primary cultures of CF lung epithelial cells. This is a classic marker for ER stress, as emphasized by the authors (Ribeiro and others 2005), and, in the case of these cultures, is associated, in part, with TLR-driven NFκB signaling and high levels of IL-8. We consider this condition to parallel the condition of our acutely isolated ex vivo CF lung epithelial cells, where we find high levels of miR-215. However, as the infection of the primary culture is gradually cured with antibiotics, Ribeiro and others (2005) find that ER stress and [Cai ++] decline. Furthermore, secreted IL-8 drops to a much lower, but still significantly elevated, level. It is possible that this reduced infection state approaches that of an infection-free culture of IB3-1, or other of the common cultured CF lung epithelial cell lines, where miR-215 is low, and the intrinsically elevated level of miR-155 is now evident. We conclude that the advantage of having a collection of miRs to study is that each miR may be targeting stability or translation of hundreds of mRNAs, allowing one to reduce the complexity of the system. However, because we need to fully understand the system, we anticipate that the next steps will include deep gene expression profiling of mRNAs as a function of exposure time and bacterial load.
miRs, inflammation, and CF
The importance of the present article rests in our delineation of miR-155 as a biomarker for uninfected and of miR-215 as a biomarker for infected human CF lung epithelial cells. Importantly, this is a property shared by both the model CF lung epithelial IB3-1 cells and ex vivo CF lung epithelial cells. These molecular biomarkers, representing hundreds of affected mRNAs, also indicate that these human CF cells carry with them an intrinsic proinflammatory defect in the uninfected state. The high level of miR-155 suppresses SHIP1, allowing the PI3K-derived inositol trisphosphate product to drive Akt, stimulate MAPKs, and stabilize the IL-8 mRNA. To this state, we find that infection induces an entirely different process, driven by miR-215, which is p53 centric, and may compromise CF lung epithelial cells when confronted with PA, or bacterial cell products. These data may also contribute to resolving the ongoing controversy over whether CF is associated with an intrinsic proinflammatory defect, or with an infection-elicited defect. In the case of human CF cells, the answer, which may satisfy both sides of this controversy, is both, if infection is also present.
Footnotes
Acknowledgments
The authors thank Uma Sajjan (Ann Arbor, Michigan) for the generous gift of the mucoid PA strains. This study was supported by USU-Intramural Funds (R.B.) and the Cystic Fibrosis Foundation (R.B. and H.B.P.).
Disclaimer
The views expressed are those of the authors and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences, the Department of the Defense, or the U.S. government.
Author Disclosure Statement
No competing financial interests exist.