
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) has caused one of the most economically devastating and pandemic diseases of swine. Previous studies have documented that PRRSV nonstructural protein-1α (nsp1α) was an interferon antagonist, but the mechanism by which nsp1α inhibited the interferon (IFN)-β production was unclear. Here, by site-directed mutagenesis of the predicted zinc-coordinating residues of the zinc-finger (ZF) domain of nsp1α or by deletion of the ZF domain of nsp1α, we explored whether the ZF domain was required for nsp1α to disrupt the IFN-β production. The results showed that both mutagenesis of the predicted zinc-coordinating residues of the ZF domain and deletion of the ZF domain made nsp1α lose its interferon antagonism activity. In conclusion, our present work indicated that the ZF domain of nsp1α was necessary for nsp1α to inhibit the IFN-β induction.
Introduction
Type I interferon (IFN-α and IFN-β) is the first responder against animal virus infections (Muller and others 1994; Weber and others 2004). When a virus infects, the virus could be recognized by the pattern-recognition receptors (PRRs) such as membrane-bound Toll-like receptors (TLRs) (including TLR3, TLR7, TLR8, and TLR9), retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs) [including the retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2)]. Among these, TLR3 and MDA5 detect the viral dsRNA, and then, TLR3 recruits the adaptor molecule TRIF (Toll/IL-1 receptor domain-containing adaptor-inducing IFN), while MDA5 recruits VISA (virus-induced signaling adapter; also known as CARDIF, IPS-1, or MAVS) to induce activation (phosphorylation) of IFN regulatory factor 3 (IRF-3) and finally induces IFN-β transcription. IRF-3 is a constitutively expressed, and latent transcription factor for the transcription of IFN-β and the activation of IRF-3 needs its phosphorylation by 2 noncanonical IκB kinases, TANK-binding kinase 1 (TBK1) and the noncanonical IκB kinases (IKK-ɛ) (Bowie and Unterholzner 2008). Then, IFN-β binds to the IFN-α/β receptor and induces the IFN-regulated genes responsible for the antiviral response (Sadler and Williams 2008). However, during the coevolution with the host cells, many viruses have developed defensive mechanisms to inhibit IFN-β production, making it difficult for host cells to defeat viral infection (Weber and others 2004; Bowie and Unterholzner 2008).
The PRRSV genome has 9 open reading frames (ORFs) composed of ORF1a, ORF1b, ORF2a, ORF2b, and ORF3–7. ORF1a and ORF1b could produce 14 nonstructural proteins (nsp1α, nsp1β, nsp2, nsp3, nsp4, nsp5, nsp6, nsp7α, nsp7β, nsp8, nsp9, nsp10, nsp11, and nap12) (den Boon and others 1991, 1995; Chen and others 2010; Shi and others 2011a). Moreover, these nsps had been screened for type I IFN suppression, and 4 nsps were identified to contain the suppressive activities: nsp1α, nsp2, nsp4, and nsp11 (Kim and others 2010). nsp2 is a membrane-anchored protein that contains a 3C-like cysteine protease activity that inhibited IRF3 phosphorylation and nuclear translocation (Li and others 2010). nsp11 contains a domain designated the NendoU domain and functions as an endoribonuclease, which was essential for nsp11 as the IFN antagonist (Shi and others 2011a), while nsp1α possessed 3 parts: an amino-terminal zinc-finger (ZF) domain [1–65 amino acids (aa)], a papain-like cysteine protease (PCP)-α domain (66–166 aa), and a C-terminal extension (CTE) (167–180 aa) (Sun and others 2009), and previous studies showed that the nsp1α PCP-α domain and CTE were essential for nsp1α to antagonize the IFN-β induction (Song and others 2010; Shi and others 2011b). However, whether the ZF domain was essential for nsp1α as the antagonist of IFN-β production was unclear. Therefore, the aim of our present work was to explore whether the ZF domain was essential for nsp1α to disrupt the IFN-β production.
Materials and Methods
Cell and virus
MARC-145 cell, a fetal green monkey fibroblast cell line derived from MA-104 (Kim and others 1993), was maintained in the Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Hyclone). PRRSV strain BJ-4 was a kind gift from Dr. Hanchun Yang (China Agricultural University).
Plasmids
The plasmids pcDNA3.1-VISA, pcDNA3.1-TRIF, p-284 Luc (positions −284 and +19 in the IFN-β gene before the luciferase report gene), pcDNA3.1-FLAG, pcDNA3.1- FLAG -nsp1α, and phRL-TK have already been described (Shi and others 2010, 2011b). Full-length GFP cDNA was amplified by using pEGFP-C1 (Clontech) as a template and was digested with NheI and HindIII, and then cloned into pcDNA3.1. The nsp1α ZF domain mutants, nsp1αCys-8→Ala (nsp1αC8A) and nsp1αC10A, were constructed by a standard PCR method using the primers that the forward primers included the mutation bases. While nsp1αC25A and nsp1αC28A mutants were constructed by using the overlap PCR, and the mutation bases were in the primers nsp1αC25A forward, nsp1αC25A reverse, nsp1αC28A forward, and nsp1αC28A reverse. To generate a GFP-tagged nsp1α ZF domain, the ZF domain cDNA was digested with HindIII and EcoRI and then cloned into pcDNA3.1-GFP. All primers used in our present work are listed in the Table 1. p55C1B Luc (Yoneyama and others 1996, 2004; Devaraj and others 2007), a firefly luciferase reporter gene plasmid containing repetitive pIRF-3-binding sites, was kindly provided by Dr. Takashi Fujita. pcDNA3-FLAG-IKK-ɛ was kindly provided by Dr. Katherine A. Fitzgerald (Fitzgerald and others 2003). All constructs were fully sequenced.
Luciferase assay
All newly prepared plasmids were verified by sequencing. Lipofectamine2000 (Invitrogen) was used for transient transfection. Cells grown in 24-well plates were transfected in triplicate with the p-284 Luc plasmid (200 ng) or p55C1B Luc plasmid (500 ng), phRL-TK (80 ng), and expression vector (600 ng) or supplied with an equivalent control vector. At the appointed time, cells were harvested, and the luciferase activity was measured using the dual luciferase reporter assay system (Promega) in the MicroBeta® TriLux liquid scintillation and luminescence counters (Microbeta-1450: Wallac).
Cell extracts and immunoblotting
MARC-145 cells grown in 24-well plates were transfected in triplicate with the various expression plasmids, and 48h later, transfected cells were lysed in a lysing buffer [1% Nonidet P-40, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM Na3VO4, 2 mM NaF, and a protease inhibitor cocktail]. The detailed procedure for immunoblots has been described (Shi and others 2008). Briefly, the total protein concentration was quantified with the Bradford protein assay (Biocolor Bioscience & Technology Company). Equal proteins were separated on SDS–polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membranes (Millipore Company), and then probed with anti-FLAG (Sigma F1804) or anti-GFP (Clontech). Proteins were detected by using an ECL detection system (Cell Signaling Technology).
Statistical analysis
Student's t-test was used for statistical analyses, and the comparisons were considered as statistically significant when P<0.05.
Results
The mutant that deleted the ZF domain in nsp1α failed to inhibit the induction of IFN-β
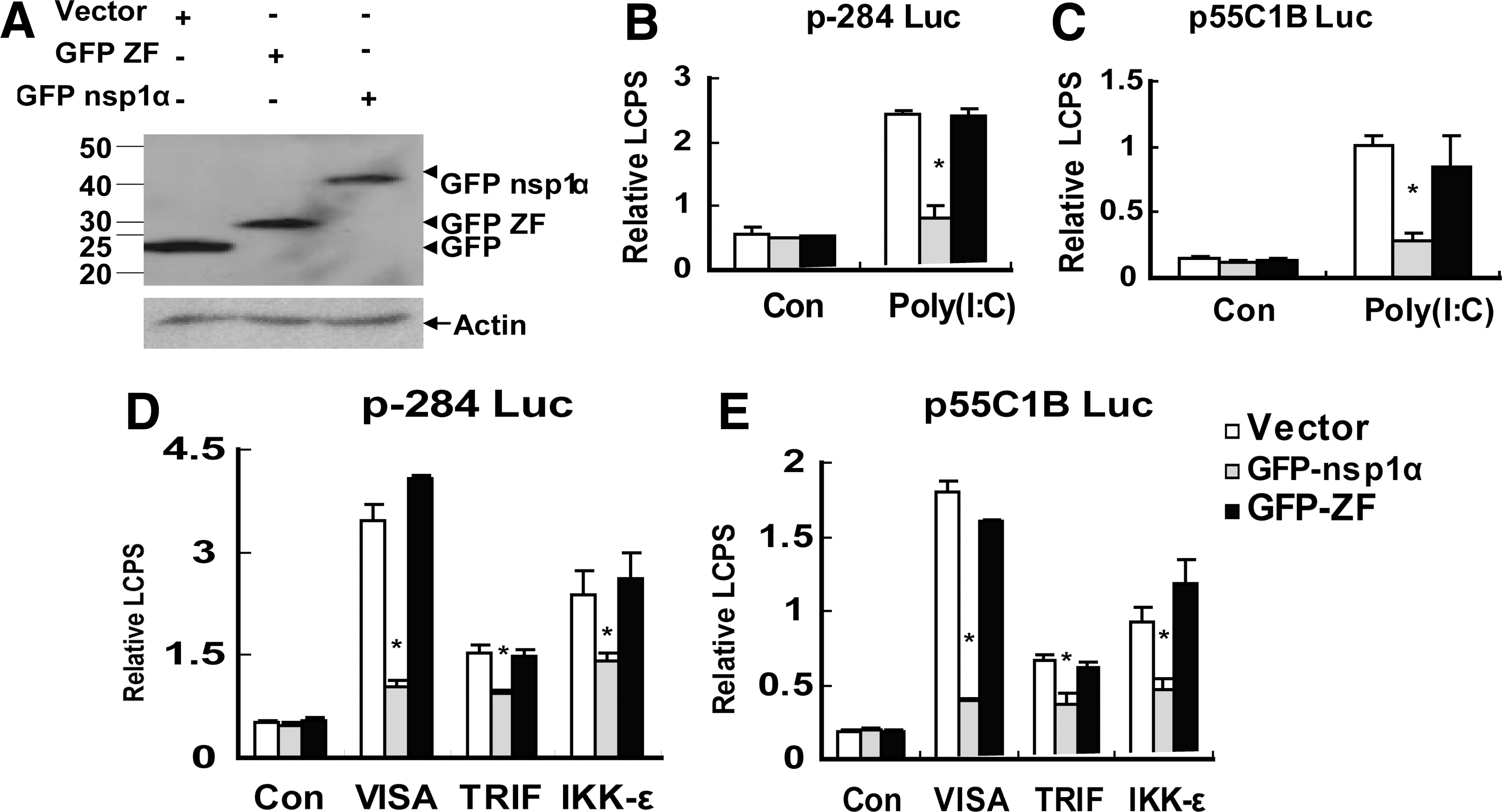
nsp1α contained 3 parts: the N-terminal ZF domain (Met1-Glu65), the PCP domain (PCPα domain, Pro66 to Gln166), and the C-terminal extension (CTE; Arg167 to Met180) (Sun and others 2009). Previous Studies have demonstrated that nsp1α inhibited the production of IFN-β (Chen and others 2010; Shi and others 2011b). To explore whether the ZF domain was essential for nsp1α as the antagonist to the IFN-β production, we deleted the ZF domain in nsp1α and constructed the expression plasmid—pcDNA3.1-FLAG nsp1α 66–180 (nsp1α DZF)—the expressions of which were confirmed by western blot (Fig. 1A), and found that the mutant that deleted the ZF domain in nsp1α failed to block Poly (I:C)(a synthetic dsRNA analog)-induced activation of the IFN-β promoter (Fig. 1B).
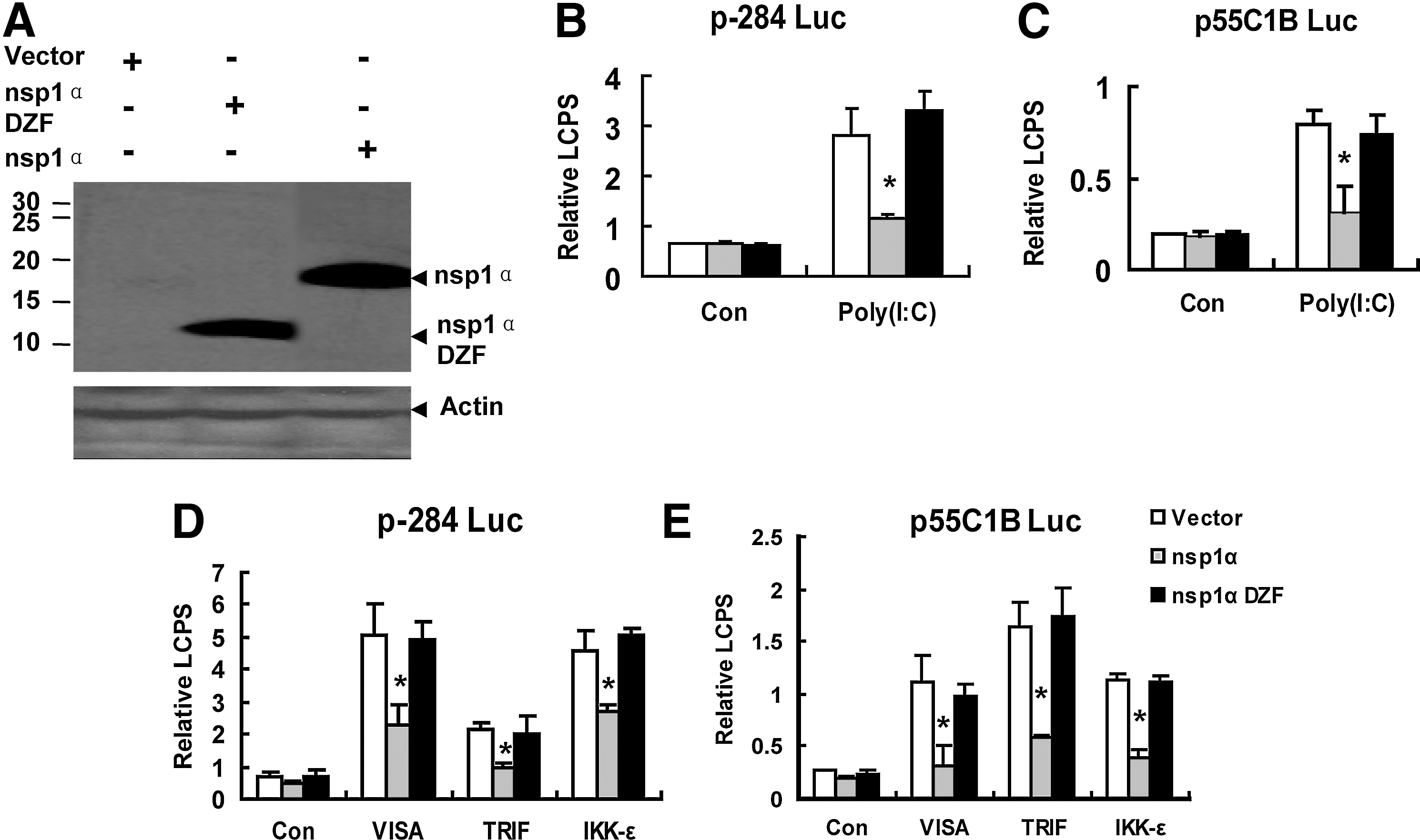
The nsp1α mutant that deleted the zinc-finger (ZF) domain failed to inhibit the activities of the interferon (IFN)-β promoter (p-284 Luc) and the pIRF-3-dependent promoter (p55C1B Luc).
Because the pIRF-3 was a necessary component to the activation of the IFN-β promoter (Peters and others 2002), p55C1B-Luc (Yoneyama and others 1998, 2004; Devaraj and others 2007), the pIRF-3-dependent synthetic promoter, was detected after the Poly (I:C) treatment or the mock treatment. As shown in Fig. 1C, nsp1α 66–180 (nsp1α DZF) could not inhibit the activation of p55C1B-Luc; that is, the results in Fig. 1C confirmed that in Fig. 1B.
Poly (I:C), a double-stranded RNA, could be recognized by TLR3 (Yamamoto and others 2003) and MDA5 (Gitlin and others 2006; Kato and others 2006; Onoguchi and others 2011). Then, through TBK1 and IKK-ɛ, TLR3 recruited TRIF, and MDA5 recruited VISA, to phosphorylate IRF-3, and finally activate the IFN-β promoter (Bowie and Unterholzner 2008). Overexpression of VISA, TRIF, or IKK-ɛ could induce the activation of IRF-3 and activate the IFN-β promoter (Yoneyama and others 2004; Devaraj and others 2007; Zhong and others 2008). Our previous study has shown that nsp1α inhibited the IFN-β production induced by overexpression of VISA, TRIF, or IKK-ɛ (Shi and others 2010, 2011b), so we investigated whether deleting the ZF domain could also influence the nsp1α to inhibit the IFN-β production induced by overexpression of VISA, TRIF, or IKK-ɛ. The results showed that the mutation that deleted the ZF domain in nsp1α could not suppress IFN-β promoter activation induced by ectopic expression of VISA, TRIF, or IKK-ɛ (Fig. 1D). Similar results were also obtained when p55C1B Luc was in place of p-284 Luc (Fig. 1E).
Mutations that mutagenesis of the predicted zinc-coordinating residues of the ZF domain of nsp1α could not inhibit the induction of IFN-β
The crystal structure of PRRSV nsp1a documented that the ZF domain of PRRSV nsp1α belonged to the 4-Cys ZF superfamily (Tijms and others 2001; Sun and others 2009) and the Cys8, Cys10, Cys25, and Cys28 participated in binding with the zinc ion (Sun and others 2009) (Fig. 2A).
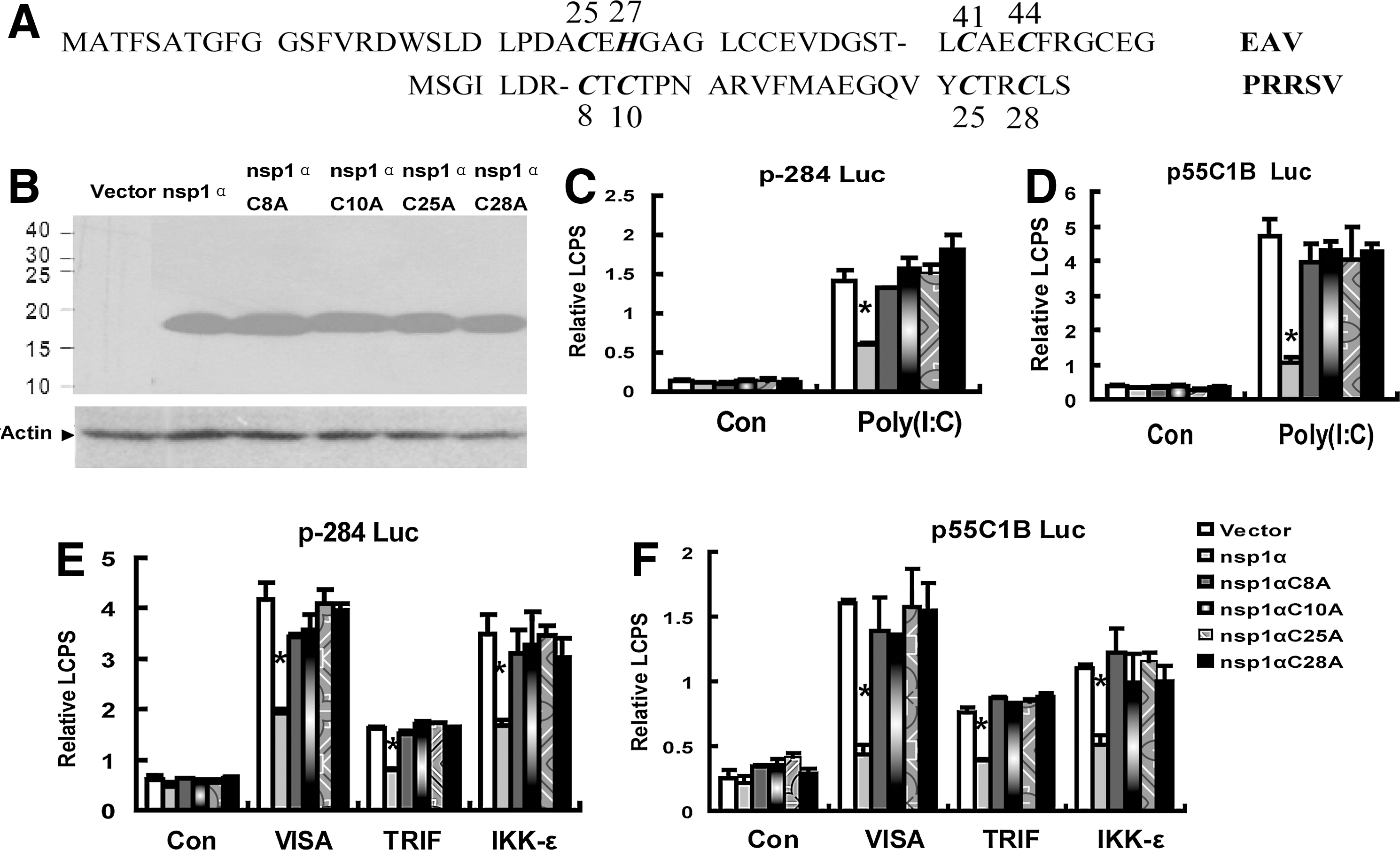
The nsp1α mutants that mutagenesis of the predicted zinc-coordinating residues of the ZF domain failed to inhibit the activities of the IFN-β promoter (p-284 Luc) and the pIRF-3-dependent promoter (p55C1B Luc).
The sequence blast showed that the ZF domain in the Arteriviridae viruses, which included PRRSV, mouse lactate dehydrogenase-elevating virus, equine arteritis virus (EAV), and simian hemorrhagic fever virus, was conserved (Tijms and others 2007; Sun and others 2009). Moreover, the ZF domain in nsp1 of EAV, another arteritis virus, played important role for the transcription of the EAV; that is, mutation in the zinc-binding residues (Cys25, His27, Cys41, and Cys44 in Fig. 2A) of the ZF domain influenced the transcription of the EAV. Therefore, next, we made single-amino-acid substitutions into the zinc-coordinating residues of the ZF domain and investigated whether the mutants could inhibit the induction of IFN-β or not. Fig. 2C shows that any of the mutant nsp1αCys-8→Ala (nsp1αC8A), nsp1α C10A, nsp1α C25A, and nsp1α C28A failed to inhibit IFN-β promoter activation induced by Poly (I:C) or the ectopic expression of VISA, TRIF, or IKK-ɛ (Fig. 2E), and the results in Fig. 2D and F also confirmed that in Fig. 2C and E.
The ZF domain of nsp1α was not sufficient to inhibit the induction of IFN-β
Since the ZF domain only contained 65 amino acids, and it was difficult to detect by western blot, we fused the ZF domain to a new tag protein GFP, the molecular weight of which was about 25 kiloDalton (kDa). We constructed the expression plasmid pcDNA3.1-GFP- ZF, and the protein expression was confirmed by the western blot (Fig. 3A), and also the GFP-nsp1α and the GFP-ZF had the same location in the cells (data not shown): both nsp1α and nsp1α ZF mainly localized in the nucleus, but also some of them retained in the cytoplasm; a scattered punctate dot-like fluorescent-staining pattern was consistently observed, and these results were similar to the previous report about the location of PRRSV nsp1α (Chen and others 2010). Next, we tested whether only the ZF domain itself could inhibit the induction of IFN-β. The results in Fig. 3 show that the ZF domain itself could not inhibit the Poly (I:C) or the ectopic expression of VISA-, TRIF-, or IKK-ɛ-induced activation of the IFN-β promoter (Fig. 3B, D) and the Poly (I:C) or the ectopic expression of VISA-, TRIF-, or IKK-ɛ-induced activation of the pIRF-3-dependent synthetic promoter (Fig. 3C, E).
The ZF domain of nsp1α was not sufficient to inhibit the activations of the IFN-β promoter (p-284 Luc) and the pIRF-3-dependent promoter (p55C1B Luc).
Discussion
PRRS is a disease of pigs that is characterized by negligible induction of type I IFNs and viral persistence for an extended period (Sun and others 2012). Previous studies have shown that nsp1α inhibited the production of IFN-β by suppression of NF-κB and inhibition of the phophosphorylation of IRF-3 in the absence of other genes (Chen and others 2010; Shi and others 2010, 2011b; Song and others 2010). However, it remains to be determined exactly how nsp1α antagonized the production of IFN-β. Since the recombinant IFN-β not only protected swine alveolar macrophages and MARC-145 cells from infection with PRRSV (Overend and others 2007), but also could reduce the yield of PRRSV in vivo (Buddaert and others 1998), identifying the viral components that inhibited the production of IFN-β, and their mechanism of this action was important. PRRSV nsp1α had 2 ZF motifs: one motif (ZF1) of C8–C10–C25–C28 and another motif (ZF2) of C70–C76-H146-M180. Sun and others (2012) considered that the PCPα activity was self-inactivated by conformational changes, and thus was less likely responsible for suppression of the IFN response and suggested that 2 ZF motifs may be associated with nsp1α-mediated inhibition of IFN-β, although our previous work shows that the PCPα activity was essential for nsp1 as the IFN-β antagonist (Shi and others 2011b). Our recent work shows that deletion of the M180 did not influence nsp1α to inhibit the induction of IFN-β (Shi and others 2012), which indicated that ZF2 may not take part in the inhibition of IFN-β, while our present results, especially which were obtained from the mutants of the putative zinc-binding amino acid of the ZF domain, provided the first evidence that the ZF domain was necessary for nsp1α to inhibit the production of IFN-β (Fig. 1 and Fig. 2). Moreover, we also found that only the ZF domain itself was not sufficient to inhibit the production of IFN-β (Fig. 3), which suggested that the ZF domain in nsp1α was not the only region participated in the inhibition of IFN-β production.
nsp1α contained 3 parts: the N-terminal ZF domain (Met1-Glu65), the PCP domain (PCPα domain, Pro66 to Gln166), and the C-terminal extension (CTE; Arg167 to Met180) (Sun and others 2009). Our and other previous studies have shown that both the activity of the PCPα and the region of the CTE were required for nsp1α to inhibit the production of IFN-β (Song and others 2010; Shi and others 2011b, 2012), while our present work gave the new information that the N-terminal ZF domain was also essential for nsp1α to inhibit the production of IFN-β. The above results indicated that all the 3 parts participated in the function of nsp1α to inhibit the production of IFN-β; that is, if there is no help of any part, the nsp1α will not inhibit the production of IFN-β, which indicated that it was possible that all the 3 parts determined the structure of nsp1α or influenced the interaction of nsp1α with the cellular proteins or the cellular components.
The ZF domain, which was one of most prevalent protein domains, was always short and was independently folded by some amino acids that needed the coordination with the zinc atom to stabilize its structure (Laity and others 2001). Moreover, the ZF domain was the common motif that participated in the interaction between protein and DNA (Klug 2010), protein and RNA (Lu and others 2003), and protein and protein (Liew and others 2005), so it was not suppressing that many viruses encoded ZF proteins that help virus antagonize the cell antiviral response. For example, the ZF domain in the infected cell protein 0 (bICP0) encoded by Bovine herpesvirus 1 (BHV-1) was essential for inhibiting the IFN-dependent transcription (Lin and others 2004; Henderson and others 2005; Saira and others 2007), and the ZF domain in EAV nsp1 was essential for the transcription of EAV (Tijms and others 2007). Therefore, our present work provided another evidence that the nsp1α ZF domain encoded by PRRSV was essential for nsp1α to inhibit the cellular antiviral response. However, the precise mechanism that the ZF domain was essential for nsp1α to inhibit the production of IFN-β warranted further investigation.
In conclusion, our present work demonstrated that the ZF domain was essential for PRRSV nsp1α to inhibit the IFN-β induction. Mutation, that is, mutagenesis of the predicted zinc-coordinating residues of the ZF domain made nsp1α lose its interferon antagonism activity. Along with the previous studies that the recombinant IFN-β not only protected swine alveolar macrophages and MARC-145 cells from infection with PRRSV (Overend and others 2007) but also could reduce the yield of PRRSV in vivo (Buddaert and others 1998), the ZF domain of nsp1α may be an exciting target for exploiting new drugs for PRRSV treatment or for choosing the modified live-virus PRRSV vaccine development.
Footnotes
Acknowledgments
We thank Takashi Fujita, Rongtuan Lin, and Katherine A. Fitzgerald for providing reagents. We also thank Ying Zhang (Henan BEBON International Co., Ltd.) for language editing. This work was supported by the Key Program National Natural Science Foundation of China (grant nos. 30730068, 31072122, and 31272546).
Author Disclosure Statement
No competing financial interests exist.