
Abstract
The interferon (IFN) family of cytokines regulates many cellular processes, such as transcription, translation, post-translational modifications, and protein degradation. IFNs induce growth inhibition and/or cell death, depending on the cell type, by employing different proteins. This review describes a novel growth-suppressive pathway employed by IFNs that affects rRNA levels. Maturation of rRNA involves numerous noncoding small regulatory RNA-guided processes. These regulatory RNAs, called small nucleolar RNA (snoRNAs), function as a ribonucleoprotein particle (RNP) in the nucleolus. The biogenesis of snoRNPs is dependent on core protein and assembly factors. Our laboratory recently isolated a growth-suppressive protein gene associated with retinoid-IFN-induced mortality (GRIM)-1 using a genetic screen. IFN-inducible GRIM-1 (SHQ1) is an assembly factor that controls one arm of the snoRNP machinery. GRIM-1 inhibits sno/scaRNP formation to induce growth suppression via reduction in mature rRNA levels. Loss of GRIM-1 observed in certain cancers implicates it to be a novel tumor suppressor. Certain snoRNAs have been reported to act as either oncogenes or tumor suppressors in vitro. Recent studies have shown that certain sno/scaRNAs are further processed into micro RNA-like molecules to control translation of protein-coding RNAs. We present a model as to how these small regulatory RNAs influence cell growth and a potential role for GRIM-1 in this process.
Introduction
T
All life forms utilize modified nucleotides to regulate their nucleic acids. In prokaryotes, methylated nucleobases (mN) in DNA by the host restriction–modification system protects their genetic material, while destroying the genetic material of the invading phage (Marinus and Morris 1973; Wilson 1991). A eukaryotic parallel of this phenomenon observed in human–mouse cell hybrids (Ruddle and Creagan 1975) helped in the initial mapping of IFN genes in humans (Creagan and others 1975). A role of this modification in eukaryotic mRNA have been reported recently (Bodi and others 2012; Dominissini and others 2012; Meyer and others 2012); in general, mN appears to confer protection for nucleic acids. In mammals, DNA methylation is associated with genomic imprinting and epigenetic phenomena often resulting in inhibition of transcription. In addition to DNA base modification, RNA molecules harbor other uniquely modified residues. It is interesting to note that certain RNA modifications show a high degree of conservation at certain positions in an rRNA species (Ofengand and Bakin 1997); and other newer modifications have been acquired depending on organismal complexity. A typical eukaryotic transcript pool consists of protein-coding RNAs and noncoding RNAs; the latter comprising ∼95% of the bulk. The noncoding RNA pool is further categorized into functional classes as follows: rRNA, tRNA, snRNA, small nucleolar RNA (snoRNA), small Cajal body RNA (scaRNA), TERC, micro RNA (miRNA), and other emerging noncoding RNAs (such as eRNA, gRNA, piRNA, saRNA, and tiRNA). A balance among these RNAs is maintained in cells at a steady state that gets disturbed under conditions like stress, inflammation, cancer, and other diseases. The 4 rRNA species (28S, 18S, 5.8S, and 5S) form the main structural scaffold of a ribosome onto which ribosomal proteins are loaded forming the mature particle that translates mRNAs in the cytoplasm. The entire maturation of rRNA takes place in the nucleolus aided by myriad of factors (∼200) that are not part of the mature ribosome (Kiss and others 2006; Terns and Terns 2006). Three major types of modified residues (mN, Nm, Ψ) are found in the mature rRNA. It is believed that these modifications are necessary for rRNA maturation and in some way helps ribosome assembly and function (Decatur and Fournier 2002). Of these, Nm and Ψ are sequence-guided processes; while the basis for mN remains obscure, even though the modifying methylase has been identified. Gross inhibition of these modifications has a detrimental effect on cells (Decatur and Fournier 2003). However, the actual role(s) of these modifications are still unclear largely owing to a lack of specific and/or direct assays. The focus of this review will be on how rRNA maturation is affected by IFNs, with a specific discussion on how sno/scaRNAs influence the cellular function in conjunction with their associated protein machinery. We also describe a RNA regulatory mechanism targeted by IFNs through a novel gene product called, GRIM1 (SHQ1), isolated as a growth-suppressive gene product in a genetic screen employed by our laboratory (Hofmann and others 2010). The yeast orthologue of GRIM1, Shq1, is necessary for the biogenesis of H/ACA RNAs and rRNA maturation (Yang and others 2002).
A Preview on snoRNAs and snoRNPs
Among the several noncoding RNAs that control cell growth are the snoRNAs represented by 2 structural classes viz., C/D and H/ACA RNAs. More than 200 snoRNAs have been identified in mammals with a wide range in size (60–300 nucleotides). All snoRNAs are derived from RNApol-II activity with a majority processed from intron(s) of certain protein-coding and noncoding transcripts, while some are transcribed independently. A few examples are listed in Table 1 to show the diversity of their role in rRNA maturation along with their location in the host gene. A comprehensive list of species-wise snoRNAs and their targets can be found at
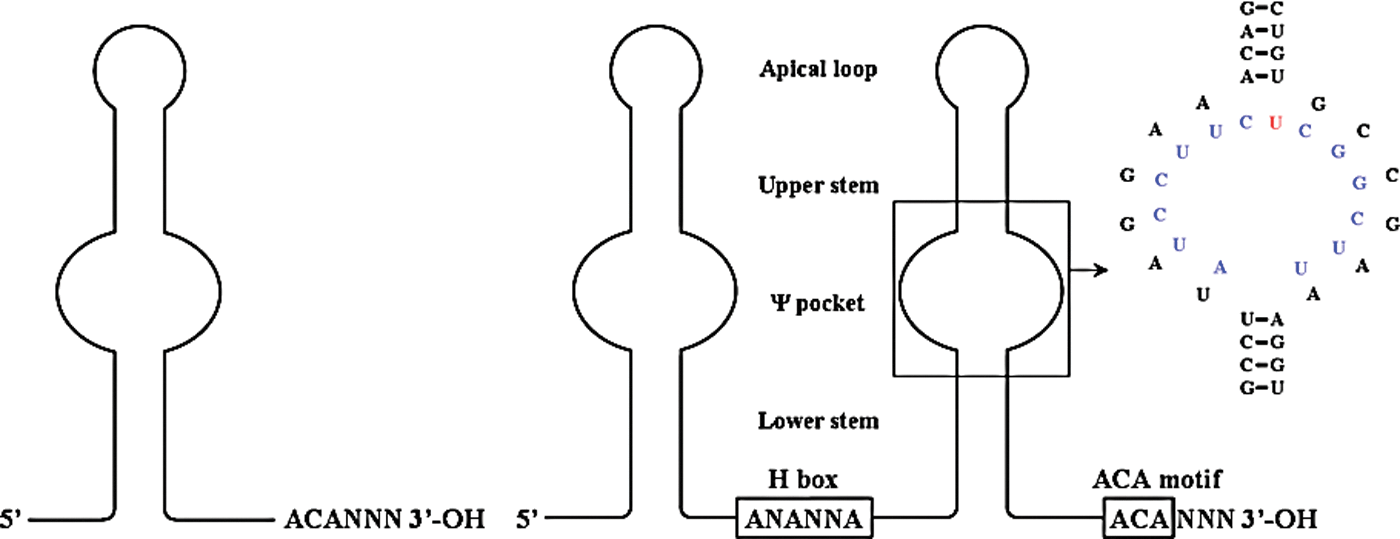
A planar representation of H/ACA RNA. These RNAs can have single or double hairpin structures. The central bulge (Ψ pocket) hybridizes to complementary sequences in rRNA (in blue) and the unpaired base (in red) is the uridine targeted for Ψ by DKC1. The specific example shown is SNORA65 hybridizing to 28S rRNA [redrawn from (Wu and Feigon 2007)]. Structure can be accessed from NCBI site using PDB ID code 2P89. Color images available online at
snoRNA, small nucleolar RNA; scaRNA, small Cajal body RNA.
Formation of a snoRNP
A tertiary structural feature in the intronic portion of the hnRNA is believed to be responsible for loading the protein(s) that specifically recognize the snoRNA region. There are some differences between C/D and H/ACA RNAs during the ribonucleoprotein particle (RNP) assembly. After the intronic region has been spliced completely, other factors (exonucleases) trim the flanking intronic sequence, thus leaving the snoRNA region intact with its bound protein complex. The protein complex for nascent and functional C/D RNP consist of Fibrillarin (FBL, an RNA methylase), Nop56, Nop58, and the kink-turn motif is recognized by NHP2L1 (NHPX, 15.5-kDa protein) (Tyc and Steitz 1989). In H/ACA RNA, the apical loop and upper stem region are recognized by NHP2 and the nascent RNP consists of Dyskerin (DKC1, the pseudouridylase), NOP10, and NAF1; the premature H/ACA RNP is devoid of NAF1 and GAR1 is incorporated to form the functional H/ACA RNP (Balakin and others 1996). The above processes occur in the nucleoplasm. Using pure components (protein and RNA), it is easier to assemble an archeal snoRNP in vitro, while it has been challenging to assemble a mammalian snoRNP.
Factors involved in snoRNP maturation
In addition to the proteins present in the mature snoRNP, other structural (Nopp140 (Isaac and others 1998), SMN (Pellizzoni and others 2001) etc.), enzymatic (RUVBL1, RUVBL2 (King and others 2001) etc.), transport (CRM1, PHAX (Boulon and others 2004) etc.) proteins are also needed for snoRNP assembly and probably maturation, although they are spatially and temporally separated from the site of snoRNA transcription and initial snoRNP formation. In addition, some assembly factors (SHQ1, NAF1, HSP90, BCD1 etc.) are needed for nascent snoRNP formation, but are not found in the later stages.
Function of snoRNPs
Transcription, processing, and maturation of rRNA occurs in the nucleolus with the aid of numerous snoRNPs, ribosomal proteins, and other factors. In addition, 5S rRNA (RNApol-III transcribed and processed outside the nucleolus) is imported into the nucleolus to be assembled into the ribosome (Zhang and others 2007). After mature snoRNPs have formed, they transiently accumulate in Cajal bodies (CBs) before moving to the nucleolus, where they assist in the formation, processing, and maturation of rRNA. In CBs, C/D RNPs reside longer than H/ACA RNPs and hence, it is believed that some additional processing or maturation events occur in this organelle that remain to be characterized (Richard and others 2003). Once they have been delivered to the nucleolus, they assist either in RNApol-I transcription or processing of the rRNA primary transcript (i.e., generation of mature 28S, 18S, and 5.8S rRNA molecules) or in rRNA folding by either of the 2 modifications (Nm or Ψ). Table 1 summarizes the function of few snoRNAs. Processing, modifications, and folding of rRNA are simultaneous events and so assigning a specific role to a particular site modification has not been greatly successful. Nonetheless, simultaneous blocking of multiple modifications delays or hinders rRNA maturation kinetics leading to imbalanced ribosomal subunits (60S and 40S) in the cytoplasm.
Regulation of snoRNAs in Cancer
Most of the snoRNA/RNP function has been deciphered using genetic studies of budding yeast; 5 snoRNAs considered crucial for cell viability (see Table 1) are involved in rRNA processing and not in modification of rRNA. Similarly, deletion or depletion of snoRNP proteins and their accessory factors manifests as synthetic lethality or poor growth of yeast cells, respectively (see ref (Fromont-Racine and others 2003), for a comprehensive list). In mammalian cells, RNAi-mediated depletion of snoRNP proteins also results in loss of snoRNA levels (Grozdanov and others 2009). On the contrary, cancer cells have higher levels of rRNA synthesis and protein turnover compared to their normal cells implying that the entire ribosome biogenesis pathway operates at very high rates (Belin and others 2009; Silvera and others 2010). The usefulness of some snoRNAs as prognostic indicators has also been evaluated in cancer patients using unbiased screens. However, recent studies have found that not all snoRNAs follow a similar trend in cancers and some of the protein factors are in fact downregulated. Studies performed in cell lines by ectopic expression of snoRNAs have indicated that they can act as either a tumor suppressor or an oncogene. Nonetheless, the actual biological role of a snoRNA-guided modification remains to be elucidated for developing novel therapeutics.
In some cancers, certain snoRNAs are present in high levels compared to others in the same group (C/D or H/ACA RNA). The location of such snoRNAs in frequently amplified chromosomal regions appears to be the primary cause for these observations. Forced expression of these snoRNAs (SNORD33, SNORD44, SNORD66, SNORD76, and SNORA42) in cancer cell lines is growth beneficial, while depletion retards cell growth and colony formation (Liao others 2010; Appaiah and others 2011; Mei and others 2011). Hence, these can be called oncogenic snoRNAs. An opposite trend is seen with certain other snoRNAs that are present in very low levels or the entire locus is lost due to chromosomal breakage. The expression of these snoRNAs (SNORD50, SNORD43, SNORD44, and SNORD48) appears to inhibit cancer cell growth in vitro similar to tumor suppressors (Dong and others 2008, 2009; Gee and others 2011). However, genetic deletion of homologous snoRNA in yeast is not deleterious to cell function, that is, no phenotypic score. Although yeast offers a suitable model for genetic analyses, it is a poor choice for querying the relevance of snoRNA-guided modifications in cellular function and growth as many mammalian sno/scaRNAs are absent in the yeast. Thus, these snoRNAs appear to influence cell characteristics outside their canonical pathway in cancer cells, which remains to be established. Using bioinformatics, some snoRNAs have been identified to undergo further processing and act as miRNAs (called sno-miRs) (Scott and others 2009). This area of snoRNA research is emerging as demonstrated by the presence of snoRNA-derived sequences in Ago1/2 complexes, the core proteins of RISC (Ender and others 2008). Considering that the levels of such sno-miRs are very low in a cell, deciphering the functional target(s) of such sno-miRs will be addressed in the coming years.
Among sno/scaRNAs, the role of TERC is so far one of the widely studied; it is needed for all epithelial-derived cancers to protect their aneuploid chromosomal sets. TERC is a special sno/scaRNP that does not function in the nucleolus; TERC helps TERT to synthesize the telomeric repeat sequence (Collins and Mitchell 2002). Mechanistic details of how TERC is transferred to TERT are unclear at present. Polymorphisms found in the TERC locus are associated with a few malignancies (Vulliamy and Dokal 2008). Even in the absence of a functional TERT, an alternate pathway (Bryan and others 1997), as seen in sarcomas, can maintain chromosomal ends. Moreover, chemically induced cancers have been reported in a Terc −/− mouse model and fifth and later generations of Terc −/− mice produce highly metastatic squamous cell carcinoma (Rudolph and others 1999; Bojovic and Crowe 2011). In humans, similar incidence of squamous cell carcinoma have been reported in patients with the X-linked form of dyskeratosis congenita (DC), which is due to point mutation(s) in the DKC1 gene that reduces TERC levels. DC presents clinically as a multisystem anomaly with features of bone marrow failure, abnormal skin pigmentation, leukoplakia, and increased susceptibility to cancer (Kirwan and Dokal 2008). However, other snoRNAs are not affected to the extent of TERC by the mutant DKC1 protein(s) although the biochemical mechanisms are unknown. A point mutation in the DKC1 gene (S485G) identified in pituitary adenoma (Bellodi and others 2010b) does not affect TERC levels in spite of being a less active protein. Interestingly, none of the point mutations identified so far in DC are in the catalytic region of the protein. A DKC1 isoform lacking the C-terminal 94 amino acids localizes in the cytoplasm, promotes growth, and cell adherence appears to be independent of its known function in the nucleolus (Angrisani and others 2011). Two H/ACA RNAs (SNORA36 and SNORA56) are produced during DKC1 splicing. However, the variant transcript that codes for the novel isoform does not produce SNORA56, which is the precursor for miRNA. Autosomal dominant and recessive forms of DC have been associated with point mutations in TERC/TERT/TINF2 and NHP2/NOP10, respectively (Vulliamy and Dokal 2008). Thus, in all cases mentioned above, critically shortened telomeres elicit genomic instability that finally resolves as cancer. Hypomorphic Dkc1 mutant mice present clinical features of DC from the first generation pointing to a telomere/telomerase-independent pathway of genomic instability with high incidence of spontaneous tumors (Ruggero and others 2003). Cancers of the breast, lung, and B-cell lymphomas were frequent in this genetic model. Interestingly, lymphocytes express high levels of the cytosolic DKC1 isoform (Angrisani and others 2011). On the contrary, expression of TERT immortalizes primary cell lines (Roy and others 2004) and malignant tumors develop in transgenic mouse models of E6 (from high-risk HPV16) expression in the skin (Song and others 1999). In the presence of E6 oncoprotein, the substrate specificity of E6AP (UBE3A; an E3 ubiquitin ligase) is altered leading to proteosomal degradation of ubiquitinated NFX1 (Gewin and others 2004) and p53 (Scheffner and others 1990) proteins; the former is a transcriptional repressor of TERT, while the latter is a cell cycle regulator. Thus, this mechanism actively promotes cell cycle and helps telomere length maintenance. In addition, translation of p53 mRNA is impaired under stress when DKC1 levels are low, thus mitigating the normal DNA checkpoint response (Montanaro and others 2010). This may explain to some extent why hypomorphic Dkc1 mouse embryonic fibroblasts are resistant to p53-mediated senescence (Bellodi and others 2010a). However, p53-dependent responses appear normal and only cell proliferation is severely affected in Dkc1-null liver cells that could survive for months without adverse effects (Ge and others 2010) and did not undergo immediate apoptosis as seen in Dkc1−/− embryos during development (He and others 2002).
While integration of HPV DNA is a stochastic process with no obvious preference to any genomic loci, adeno-associated viruses (AAV) prefer to integrate at a specific site, which happens to be an imprinted locus. Studies in mouse models have uncovered high-frequency integration of AAV on chromosome 12 near the Meg3 locus (Donsante and others 2007). This region is rich in snoRNA and miRNA genes and the integration event potentially affecting host gene expression has been implicated in the development of hepatocellular carcinoma. The syntenic region in humans is proposed to contain tumor suppressors. Expression of the noncoding MEG3 transcript is severely suppressed in various tumor types and tumor cell lines, while genomic deletion was associated with aggressive tumors (Zhou and others 2012). Meg3 RNA upregulates p53 levels (Zhou and others 2007), by downregulating Mdm2 levels, and promotes angiogenesis by increasing Vegfa and Vegfr1 levels (Gordon and others 2010). The adjacent Meg8 (Rian) locus produces a long noncoding RNA that is differentially expressed in developing embryonic tissues (Gu and others 2012). Silencing of Meg8 expression is needed for induced pluripotent stem cell growth (Stadtfeld and others 2010) akin to cancer cell growth. In spite of a lack of sequence conservation between human and mouse in and around MEG3-MEG8 loci, numerous snoRNAs and miRNAs are produced in this region in both species. Multiple copies of SNORD113 and SNORD114 are present only in humans, while numerous miRNAs are present in both human and mouse in this chromosomal locus. The function(s) of SNORD113 and SNORD114 are not known at present.
Cell type-specific imprinting regulates the Snurf-Snrpn-Ube3a locus in adult tissues. This locus contains multiple copies of SNORD115 and SNORD116 whose nascent RNAs have C/D RNA features with no known role on rRNA processing. Clinically, loss of the maternal allele manifests as the Angleman syndrome, while the Prader–Willi syndrome results due to loss of the paternal allele (Nicholls and Knepper 2001); autism-spectrum disorders are seen in both cases. Studies have shown that SNORD115 is processed to a smaller RNA to be incorporated into hnRNPs and assists in splicing of hnRNA to generate transcript variants and, hence, proteome diversity (Kishore and others 2010). SNORD115 is needed for the correct splicing of Htr2c (a serotonin receptor) RNA to generate a receptor with optimal ligand binding and signaling characteristics (Kishore and Stamm 2006). Studies have implicated loss of SNORD115 (de los Santos and others 2000) or SNORD116 in the Prader–Willi syndrome (Runte and others 2005; Sahoo and others 2008). Other studies in mouse models have indicated Ube3a as the candidate gene and excluded SNORD115/116 to be the cause (Jiang and others 1998). Additionally, Ube3a undergoes alternative splicing, while HTR2C mRNA is edited by ADAR2 to generate receptor diversity (Flomen and others 2004). Since, ubiquitin ligases can modulate the strength and duration of the signal by regulating ligand/receptor levels, both UBE3A isoforms and SNORD115/116 may contribute to some extent in these neurological conditions, and recently this locus has been implicated in pancreatic endocrine insufficiency (Stefan and others 2011).
Another widely studied alternatively spliced gene is PKM (Noguchi and others 1986). The M2 transcript variant is the dominant form observed in embryonic tissues, normal proliferating cells, and cancer cells (Mazurek and others 2005). Spontaneously immortalized 3Y1 fibroblasts show transcripts only for the M2 variant, while M1 variant is undetectable (Nallar and Kalvakolanu, unpublished). Transcription factors (ZFX, MYC, E2F1, STAT3 etc.) have been implicated in upregulating hnRNPs A1/A2 and PTB to suppress M1 transcript variant in cancer cell lines (Clower and others 2010) (see (Chaneton and Gottlieb 2012) for a review). What determines alternate splicing of PKM RNA in a physiological context is not known at present; however, a snoRNA-derived regulatory RNAs may be indirectly involved. It is believed that the M2 isoform helps partitioning carbon pool to sustain accelerated growth and at the same time helps cell survival when oxidative stress levels are high. The C/D snoRNAs (SNORD32A, SNORD33, and SNORD35A) produced from RPL13A introns have been suggested to regulate oxidative stress during carcinogenesis (Michel and others 2011), while RPL13A regulates translation of certain cellular mRNAs under specific conditions (Mukhopadhyay and others 2008). Interestingly, hnRNPA1 is also involved in alternate splicing of SMN2 mRNA, by antagonizing hnRNPQ1, producing a defective SMN2 protein leading to spinal muscular ataxia in a Smn1− /− mouse background (Chen and others 2008). SMN proteins are needed for maturation of snoRNPs and the exact mechanism for this tissue-specific defect is unclear. Regulation of alternate splicing by snRNA-U2 harboring noncanonical Ψ may be relevant in such scenarios (Wu and others 2011).
Recent studies indicate AID regulates certain non-IgG loci rich in repetitive sequences, such as SNHG3 or RCC1 locus (Kato and others 2012). The E1 snoRNA (U17 or SNORA73A/B) produced from this locus (Pelczar and Filipowicz 1998) undergoes uridine insertional editing. Earlier biochemical analyses had shown that E1 snoRNPs were asymmetric; multiple copies of E1 snoRNAs were present in the snoRNP along with other noncore proteins (Eliceiri 2006). The most important role of E1 snoRNP is in rRNA processing to generate the mature 18S rRNA (Mishra and Eliceiri 1997).
The spliceosome is a multicomponent RNP and all 5 snRNAs (RNUs) carry modified residues (Nm and Ψ) guided by 19 scaRNAs. Fragments identical to SCARNA3, SCARNA8, SCARNA15, and SCARNA16 have been isolated from Ago1/2 protein complexes and reported to function like miRNAs (Ender and others 2008). SCARNA15-derived miRNA-like fragment targets the Mediator subunit CDK11 mRNA, thus impacting transcription. In the same study, fragments identical to SNORA3, SNORA36B, SNORA50, and SNORA56 were also isolated. The host genes for most of these snoRNAs have a direct role in translation. Still it is unclear whether processing of these snoRNAs into miRNAs is a default cellular process. The consequence(s) of blocking such a process also needs to be addressed in the coming years. Nonetheless, it is reasonable to believe that this is used as a relay signal between the transcriptional and translational apparatus to maintain intracellular homeostasis.
Two other ncRNAs (ZFAS1 and GAS5) hosting snoRNA genes have been implicated in the development of breast cancer. ZFAS1 RNA is an antisense transcript against ZNFX1 (Askarian-Amiri and others 2011), a transcription factor, and 3 C/D RNAs (SNORD12, SNORD12B, and SNORD12C) originate from its introns and all guide Nm of a single guanine residue (3,878) in 28S rRNA. GAS5 locus produces 10 C/D RNAs that guide Nm of different residues in 18S and 28S rRNA. Reduction in GAS5 expression and SNORD44 were strongly associated with poor survival in head and neck SCC and breast cancer (Mourtada-Maarabouni and others 2009). Similarly, loss of SNORD50 expression has been reported in prostate and breast cancers (Dong and others 2008, 2009) while a genetic deletion has been reported in diffuse B cell lymphoma (Tanaka and others 2000). However, SNORA42 expression is high in non-small-cell lung cancer due to chromosomal amplification and is associated with poor prognosis (Liao and others 2010).
GRIM-1 links IFN action to snoRNA machinery
IFNs were the first cytokines to be developed into therapeutics against certain malignancies and viral diseases. Many solid tumors are refractory to IFN stimulation that can be overcome by pretreatment with retinoids. Retinoids are vitamin A-related compounds that control embryonic and postnatal growth/development in vertebrates. Deficiency of vitamin A leads to night blindness in humans, while animals show a higher cancer incidence. Earlier several preclinical and clinical studies indicated a strong synergistic growth-suppressive effect of retinoid-IFN combination on solid tumors (Kalvakolanu 2004). Using a genome-wide knockdown approach, our laboratory identified a set of genes needed for retinoid-IFN-induced mortality of cancer cells (Hofmann and others 1998; Angell and others 2000) and named them as genes associated with retinoid-IFN-induced mortality (GRIM). Blockade of expression of any individual GRIMs by an antisense construct confers protection against retinoid-IFN-induced cell death. GRIM-1 isolated by this approach is involved in regulating snoRNAs and rRNA levels (Hofmann and others 2010) (Fig. 2). In vitro translation of GRIM-1 transcript produces 3 GRIM-1 isoforms (α, β and γ) from the same open reading frame, ie, proteins differ in their N-terminal region. Whether a mammalian cell translates GRIM-1 mRNA in a similar manner is not clear at this stage. However, a polyclonal antibody recognizes bands corresponding to 3 proteins in Western blot analyses. IFN stimulation increases the levels of all isoforms and interestingly, caspase-9 cleaves GRIM-1α to generate the shorter isoforms (β and γ) that are more apoptotic than GRIM-1α; loss of mitochondrial membrane potential appears to activate caspase-9 under such a circumstance (Hofmann and others 2010) (Fig. 2). We have reported earlier that another GRIM, GRIM-19, downregulates the expression of certain antiapoptotic members of the Bcl2 family of proteins. Thus, retinoid-IFN combination can promote mitochondrial permeability transition. Overexpression of individual GRIM-1 isoforms induces growth suppression and apoptosis of cancer cell lines in a differential manner with GRIM1α and GRIM-1γ being the weakest and the strongest inducer of apoptosis, respectively. However, a moderate expression is tolerated and cells grow slower than the controls. GRIM-1 isoforms caused a significant drop in SNORA73 (U17), SNORA74 (U19), SNORA50, SCARNA8, SCARNA13, TERC, snRNA (RNU2), and SNORD3 (U3) levels with a concomitant increase in unprocessed rRNA levels appear to trigger these events (Nallar and others 2011) (Fig. 2). A converse observation was made upon GRIM-1 depletion from cells using specific shRNAs. Since the shRNA downregulates all GRIM-1 isoforms it does not definitively prove which specific isoform is the most potent inhibitor. Overexpression experiments usually have the caveat of the presence of endogenous isoforms. Hence, we reconstituted the individual RNAi-resistant GRIM-1 constructs into cells that were previously depleted of endogenous GRIM-1. These experiments showed that GRIM-1γ was the most potent suppressor of sno/scaRNA levels (Nallar and others 2011). In the presence of IFN, DKC1 levels steadily dropped over time and reach a minimum when GRIM-1 protein levels were highest (Kalvakolanu, unpublished). Surprisingly, the NAF1 protein level is unaffected in the same circumstance (Nallar and others 2011). In the presence of GRIM-1 isoforms, NAF1 was primarily found outside the nucleus, indicating that GRIM-1 alters cellular localization of NAF1. The consequence of such effects is lowering snoRNP levels below a threshold. A similar inhibitory effect of GRIM-1 (SHQ1) on H/ACA RNP assembly is evident in vitro (Grozdanov and others 2009). Depletion of GRIM-1 levels, by RNAi, conferred a growth advantage in the presence of retinoid-IFN combination and its ectopic expression induces growth suppression of cancer cell lines. Since Shq1p-depleted yeast grew at a slower rate, we tested if yeast Shq1p would act as growth promoter in the mammalian cells. Quite the contrary, upon reconstitution of yeast Shq1p into HeLa cells depleted of the endogenous GRIM-1, expression levels of sno/scaRNAs were suppressed when compared to the controls and significantly suppressed rRNA maturation (Nallar and others 2011). Thus, the functional differences in GRIM-1 action appear to be due to differences in their respective cellular environments, but not their primary sequences. Mammalian cells may have other modifying factors that control GRIM-1 activity, unlike yeast cells. Thus, GRIM-1 appears to be a novel tumor suppressor based on several independent observations: (i) the loss of GRIM-1 expression in prostate cancer specimens by immunohistochemical analysis (Nallar and others 2011), (ii) the monoallelic or biallelic loss in prostate cancers using an integrated oncogenomic approach (Taylor and others 2010), and (iii) the genomic loss in leukemias by high-resolution microarrays (Bullinger and others 2010). A prostate cancer cell line (VCaP) derived from vertebral metastases had very low levels of GRIM-1 mRNA compared to a noncancerous cell line (RWPE1) (Nallar and others 2011). That an exogenous agent (light signals focused on retina) upregulated GRIM-1 has been independently reported by another study (Schippert and others 2008) wherein the axial ocular growth was halted when chicks experienced an imposed image defocus (using external lenses).
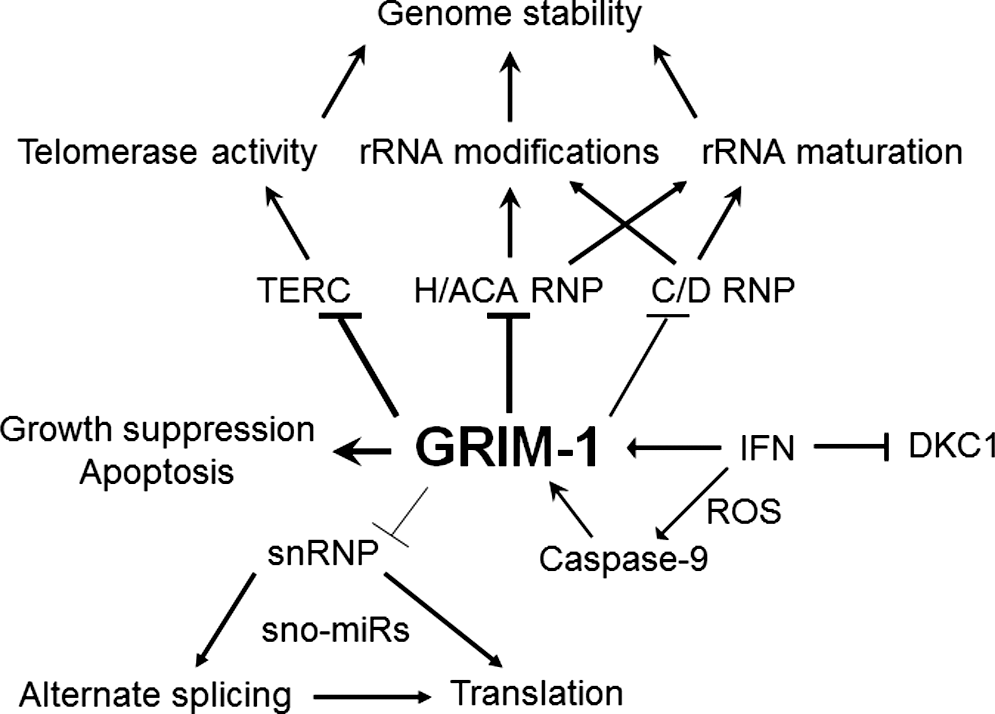
GRIM-1 is a novel tumor suppressor. Normal cells need a certain level of GRIM-1. IFN can upregulate GRIM-1 levels to cause growth suppression and/or apoptosis by 2 mechanisms: (1) by delocalization of NAF1 and (2) by downregulation of DKC1 levels. IFN-inducible GRIM-1 is cleaved to a potent apoptotic molecule by caspase-9. In the presence of high GRIM-1 levels in the cell, maturation of H/ACA and TERC RNA into RNPs is blocked (thick lines). Some C/D RNAs are also affected in the same circumstance (thin lines). All these are needed to maintain genome stability. GRIM-1 also represses certain snRNPs that are needed for spliceosome function. Blockade of spliceosome can lead to aberrant splicing. GRIM, gene associated with retinoid-IFN-induced mortality; IFN, interferon; RNP, ribonucleoprotein particle.
Isolation of several independent genes by our genetic approach suggested a network of noncanonical cell death regulators (GRIM engine) (Kalvakolanu 2004). In this model, a gene product performs a specific task that makes the cancer cell vulnerable to cell death; the strength of each gene product to induce cell death is variable. The loss of mitochondrial membrane potential and/or unregulated reactive oxygen species activate latent caspases to cleave proteins leading to cell death (Fig. 2). Whether caspase-9-mediated GRIM-1 cleavage occurs at steady state is unclear at this stage. Nonetheless, in the presence of IFN, caspase-9 cleaves GRIM-1 into a potent toxic molecule inhibiting snoRNP formation and rRNA maturation (Hofmann and others 2010).
Future perspectives
A recent study showed tissue-specific differences in snoRNA expression levels in normal adult tissues; brain tissues were rich in C/D RNAs and poor in H/ACA RNAs, while in testis, the opposite trend was observed, that is, rich in H/ACA RNAs and poor in C/D RNAs (Castle and others 2010). A few important questions arise in the light of these findings—does this also reflect in a reduction of such modified bases (Nm and Ψ) in a stoichiometric manner in a single rRNA molecule? If yes, how ribosome is fine-tuned when one of the modifications is skewed with respect to the other? Are there distinct pools of ribosomal complexes designated for a specific purpose? Nonetheless, both these snoRNA-guided modifications (Nm and Ψ) have been implicated in resistance to antibiotics as these modifications cluster near functionally important regions on the ribosome/rRNA (Rakauskaite and others 2011). Similarly, gene knockout and mutational studies have shown deficiency and mutants of ribosomal proteins display cell type-specific differences (Anderson and others 2007; Morimoto and others 2007) that prompt a new question—which ribosomal proteins are needed to form the core structural and functional modules of a mammalian ribosome in each tissue. Future studies are required to clarify the function(s) played by an individual or a set of snoRNA(s) in a tissue that normally is not enriched for the same. Similarly, more detailed genetic studies are needed for understanding the impact of the upstream regulators of sno/scaRNAs and their relevance to human cancer. Whether sno/scaRNAs and their regulators also affect the processing of other coding and noncoding RNAs remain to be investigated.
Footnotes
Acknowledgment
D.V.K. is supported by the NIH grant CA105005.
Author Disclosure Statement
No competing financial interests exist.