
Abstract
Cigarette smoking is a major established environmental risk factor for rheumatoid arthritis (RA), and synoviocyte-derived proinflammatory cytokines are implicated in the pathogenesis of RA. We have reported that aryl hydrocarbon or cigarette smoke condensate (CSC) is able to upregulate the production of proinflammatory cytokines from an RA patient-derived synovial fibroblast cell line MH7A. In this study, we compared the effect of CSC on induction of interleukin-1β (IL-1β) from RA or osteoarthritis (OA) patient-derived synovial fibroblasts, and studied the mechanism of the effect of CSC. CSC induced IL-1β mRNA from RA patient-derived synoviocytes and MH7A, but not from OA patient-derived synoviocytes. CSC induced the mRNA and both precursor and mature forms of IL-1β, and caspase-1 activity in MH7A. The mechanism of CSC-induced IL-1β mRNA expression was investigated in MH7A. Reporter gene analyses and promoter pull-down assay indicated that 3 novel NF-κB sites at −3771 to −3762 bp, −3105 to −3096 bp, and −2787 to −2778 bp in the promoter region of the IL-1β gene, especially the far distal NF-κB site and NF-κB activation, are critical for the gene activation by CSC. CSC-induced NF-κB activation, IL-1β promoter activity, IL-1β mRNA upregulation, and CYP1A1 mRNA induction were all inhibited by an aryl hydrocarbon receptor (AhR) antagonist α-naphthoflavone. These results indicate that CSC induced IL-1β production from RA patient-derived synoviocytes, but not OA patient-derived synoviocytes, through AhR-dependent NF-κB activation and novel NF-κB sites.
Introduction
Epidemiological studies indicate an association of cigarette smoking with disease outcome in patients with early inflammatory polyarthritis (Harrison and others 2001), the increase of rheumatoid factor, and nodule formation in patients with RA (Tuomi and others 1990), and a strong association was observed between heavy cigarette smoking and RA, particularly in patients without a family history of RA (Hutchinson and others 2001). The risk of smoking for the disease becomes extremely high in individuals, either men or women, with shared epitope in HLA-DRB1, which is a major genetic risk factor for RA (Padyukov and others 2004). Maternal smoking in pregnancy is also a determinable factor of infant RA and other inflammatory polyarthritis (Jaakkola and Gissler 2005). However, the experimental bases supporting the epidemiological studies are very few (Onozaki 2009).
We have previously reported that polycyclic aromatic hydrocarbons (PAHs), the constituent of cigarette smoke such as 3-methylcholanthrene (3-MC), benzo[a]pyrene (B[a]P), and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), upregulated IL-1β mRNA in RA patient-derived SV40 T antigen-transformed human synovial fibroblast cell line MH7A (Tamaki and others 2004), which has similar characteristics as parental synoviocytes (Miyazawa and others 1998a, 1998b). We also reported that cigarette smoke condensate (CSC), either mainstream or side stream, also induced IL-1α, IL-1β, IL-6, and IL-8 at both mRNA and protein levels in MH7A (Shizu and others 2008). In this study, we compared the responsiveness of MH7A, RA-patient derived primary synoviocytes, and osteoarthritis (OA) patient-derived synoviocytes to CSC, and investigated the mechanism of the effect of CSC. We showed that CSC induced IL-1β mRNA from MH7A and RA patient-derived synoviocytes, but not OA patient-derived synoviocytes. We also showed that CSC induced IL-1β mRNA through aryl hydrocarbon receptor (AhR)-dependent NF-κB activation and novel NF-κB sites in the IL-1β promoter region.
Materials and Methods
Reagents
RPMI1640, Dulbecco's modified Eagle's medium (DMEM), B[a]P, α-naphthoflavone (α-NF), and polymyxin B were purchased from Sigma-Aldrich Co. Fetal bovine serum (FBS) was purchased from Hyclone Laboratories, Inc. Human recombinant TNF-α was provided by Dainippon Pharmaceutical Co. The specific activity of TNF-α was 6×107 U/mg based on the cytotoxic assay using L929 cells cultured in the presence of actinomycin D (Ruff and Gifford 1981).
Preparation of CSC
CSC was prepared as described previously (Kamiya and others 2005). A common American brand of cigarette was used in this study. Each cigarette was 84-mm long, 25 mm in circumference, and had a charcoal filter that adsorbs normally 9 mg of tar, and 0.7 mg of nicotine. Particulate matters from mainstream were collected using cigarette smoke collection apparatus as described previously with several modifications (Grimmer and others 1987). The yield of mainstream CSC was 9.25 mg per cigarette. The extract was dissolved in ethanol and used as CSC.
Cell culture
MH7A is an immortalized cell line obtained by stably transfecting rheumatoid fibroblast synoviocyte cells with the SV40 T antigen gene. MH7A expresses IL-1 receptor, intercellular adhesion molecule-1 (ICAM-1), CD16, CD40, CD80, and CD95. IL-1β enhanced the production of IL-6 and stromelysin-1, and the surface expression of ICAM-1, in a manner similar to that in the parental cells (Miyazawa and others 1998a 1998b). MH7A cells were cultured in RPMI1640, 100 U/mL of penicillin G, 100 μg/mL of streptomycin, 15 mM HEPES, and 10% heat-inactivated FBS at 37°C. Primary synovial fibroblasts from RA patients or OA patients were cultured in the DMEM, with 100 U/mL of penicillin G, 100 μg/mL of streptomycin, 4 mM
RNA preparation and reverse transcriptase reaction
Total RNA from cells was extracted as described previously (Matsumura and others 2000). The amount of RNA was determined by spectrophotometrically. The RNA was reverse transcribed by incubating 2 μg of total RNA in a 20-μL reaction volume containing 50 mM Tris–HCl (pH 8.3), 40 mM KCl, 6 mM MgCl2, 10 mM DTT, 0.5 mM dNTP, 40 ng Random Primer p(dN)6, 6 U ribonuclease inhibitor, and 40 U M-MLV Reverse Transcriptase. The reaction mixtures were incubated at 37°C for 60 min, followed by 10-min incubation at 70°C in Program Temp control system PC-700 (ASTEC). The reaction mixtures were diluted 5 times with ddH2O and used as cDNA solution. cDNA samples were stored at 4°C until polymerase chain reaction (PCR) analysis.
Real-time PCR analysis
Assays were performed using an Applied Biosystems 7300 sequence detector (Applied Biosystems). For the analysis of human IL-1β and CYP1A1, each amplification mixture (20 μL) was made to contain 80 ng cDNA, 10 μL Premix EX Taq™ (pPerfect Real Time, RR039A; Takara Bio), 1 μL PCR primer (forward and reverse primer), 0.4 mL Rox Reference Dye (Takara Bio), and distilled water. The reaction mixtures were incubated at 95°C for 10 s, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, and then cooled to 4°C. For GAPDH, the reaction mixture (20 μL) was made to contain 40 ng cDNA, 10 μL POWER SYBER Green PCR Master Mix (P/N 4367659; Applied Biosystems), 2 μL PCR primer (forward and reverse primers; 5 μM), and distilled water. The reaction mixtures were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 58.2°C for 1 min, and then 60°C for 30 s, 95°C for 15 s, and then cooled to 4°C. Data were analyzed with ABI Sequence Detector's software. The results were expressed as mean±SD for the relative expression levels compared with GAPDH, and minimum values of 3 independent experiments were determined. Primers for human IL-1β (Hs00174097) and CYP1A (Hs00153120) were obtained from Applied Biosystems. Primers for human GAPDH, forward primer 5′-CCTGCACCACCAACTGCTTA-3′, and reverse primer 5′-TCTTCTGGGTGGCAGTGATG-3′ were obtained from the Japan Genetic Institute.
Plasmids
PGL3-basic, pGL4.74[hRlucTK], and pGL4.10-luc2 were purchased from Promega. pGL4-(Ig-κB)6-Luc, which contains 6 tandem repeats of Ig-κB, was generated by PCR. DNA fragments of the promoter region of the IL-1β gene were cloned into the multiple cloning site of pGL4.10-luc2 to generate pGL4.10-IL-1β (−3791), pGL4.10-IL-1β (−2371), and pGL4.10-IL-1β (−474). The pGL4.10-IL-1β mutants were constructed by deleting
Transient transfection and luciferase assays
MH7A cells were cultured in the RPMI1640 medium supplemented with 10% heat-inactivated FBS. The IL-1β reporter plasmid and pGL4.74[hRlucTK] plasmid (for normalization of transfection efficiency) were transiently transfected into MH7A cells using the calcium phosphate–DNA coprecipitation method. After 15 h of transfection, the medium was changed to the RPMI1640 medium containing 0.1% FBS, kept for 3 h, and then the cells were treated with CSC or B[a]P with other reagents for additional 24 h and harvested. The luciferase reporter gene assay kit (Roche, Germany) was used for Luciferase assay, according to the manufacturer's instructions. The light emission was measured by multilabel counter 1420 ARVO (Pharmacia). The relative luciferase activity was determined by a Dual-Luciferase® Reporter (DLR®) Assay System (Promega).
Measurement of cytokine levels
Protein level of IL-1β was determined by enzyme-linked immunosorbent assay (ELISA) Kit according to the manufacturer's protocol by using the Human IL-1β ELISA Set (BD Biosciences).
Measurement of caspase-1 activity
Caspase-1 activity in the cell lysates was determined by the Caspase-1 Fluorometric Assay Kit (R&D Systems) according to the manufacturer's protocol.
Western blot assay
After culture, the cells were washed with ice-cold phosphate-buffered saline, and lysed in an RIPA buffer [50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholate, and 1% Triton X-100] supplemented with protease inhibitors, phosphatase inhibitors, and 20 mM imidazole. After centrifugation (8,000 rpm for 5 min), the supernatant was obtained as a cytoplasmic fraction. The pellets were washed twice in an ice-cold washing buffer (20 mM HEPES-KOH, pH 8.0, 20% glycerol, 0.1 M KCl, 0.2 mM EDTA, and 0.5 mM dithiothreitol) supplemented with protease inhibitors and phosphatase inhibitors, resuspended in an RIPA buffer, and lysed by sonication and boiling. The cytoplasmic fraction was used for the analysis of IκB degradation, and cytoplasmic and nuclear fractions were used for p65 translocation. The proteins were separated by 10% SDS–polyacrylamide gel electrophoresis (PAGE) and were transferred to a polyvinylidene difluoride (PVDF) microporous membrane, Immobilon TMPVDF (Millipore). After blocking with 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20, membranes were incubated with anti-IκBα (c-15) rabbit IgG or anti-NF-κB p65(c-26) rabbit IgG (Santa Cruz Biotechnology, Inc.), anti-β-actin (AC-15), mouse mAb (Sigma Chemical Co.), and then with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Jackson ImmunoResearch) or HRP-linked anti-mouse Ig whole antibody (GE Healthcare UK, Ltd). For the analysis of IL-1β protein, the culture supernatants were concentrated with a Microcon YM10 tube (Merck Millipore), and then western blot was performed by using 15% SDS-PAGE, biotinylated anti-human IL-1β/IL-1F2 goat IgG (R&D Systems), and streptavidin–HRP conjugate (BD Biosciences). The reactive proteins were detected with enhanced chemiluminescence reagents (GE Healthcare UK, Ltd.) and were analyzed by a chemiluminescence image analyzer, LAS-1000 (GE Healthcare UK, Ltd.).
Promoter pull-down assay
By using PCR with a biotinylated primer, the IL-1β promoter fragment was biotinylated. MH7A cells were treated with or without CSC for 30 min. Cells were lysed in an RIPA buffer supplemented with protease inhibitors. The nuclear extract was obtained by the method of Ohno and others (2010). In brief, the cell lysate was centrifuged for 10 min at 20,000 g, and the pellet was resuspended in an equal volume of lysis buffer, added to an equal volume of 2 M KCl (final concentration, 1 M), and incubated for 30 min on ice. The resuspended pellet was centrifuged for 30 min at 418,600 g, and the supernatant was dialyzed with 0.1 M HM buffer containing 25 mM HEPES-KOH (pH 7.9), 0.1 M KCl, 12.5 mM MgCl2, 20% glycerol, and 1 mM DTT. The dialysate was centrifuged for 10 min at 17,400 g, and the supernatant was flash-frozen and stored at −80°C. The nuclear extract was supplemented with 0.2 mg/mL polydeoxyinosinic-deoxycytidylic acid and incubated with a 5 nM biotinylated IL-1β promoter fragment for 2 h at 4°C. Fifty milligrams of streptavidin-conjugated magnetic particles (Promega) was then added and incubated for an additional 1 h. The particles were washed 4 times, and proteins were eluted with a sample buffer and subjected to western blot analysis.
Statistic analysis
Statistical significance between 2 groups was examined using unpaired Student's t-test. The symbols * and ** indicate statistical significance at P values of 0.05 and 0.01 in figures, respectively.
Results
Differential effects of CSC on IL-1β mRNA expression in RA or OA patient-derived primary synovial fibroblasts
To examine the effect of CSC on IL-1β mRNA expression in RA or OA patient-derived primary synovial fibroblasts, cells were treated with varying doses of mainstream CSC for 24 h, and then the mRNA level of IL-1β was determined by real-time PCR. The time was chosen because the 24-h culture was maximal in induction of IL-1β mRNA by CSC in MH7A without cytotoxicity (Shizu and others 2008). TNF-α was used as a reference stimulant. Clinical details of 6 patients included in this study are shown in Table 1. As shown in Fig. 1, although there were minor differences, CSC dose-dependently upregulated the expression level of IL-1β mRNA in all the RA patient-derived synovial cells. In contrast, none of the OA patient-derived synovial cells were responsive to CSC. TNF-α tended to upregulate the IL-1β mRNA expression in the RA patient-derived synovial cells than the OA patient-derived synovial cells.

Effect of cigarette smoke condensate (CSC) on the mRNA expression of interleukin-1β (IL-1β) in rheumatoid arthritis (RA) or osteoarthritis (OA) patient-derived primary synovial fibroblasts. Cells were treated with vehicles (control) and varying concentrations of CSC or tumor necrosis factor-α (TNF-α; 5 U/mL) for 24 h. Then total RNA was extracted, and IL-1β and GAPDH mRNA expression levels were determined by real-time polymerase chain reaction (PCR). **P<0.01, *P<0.05, n.s., not significant between control and others. Mean±SD based on triplicate is shown.
OA, osteoarthritis; RA, rheumatoid arthritis; CRP, C-reactive protein.
Effects of CSC on the mRNA expression, protein induction, and gene activation of IL-1β in MH7A
As previously reported, CSC dose-dependently upregulated the expression levels of IL-1β mRNA and protein in MH7A after 24 h of stimulation (Fig. 2A, B). TNF-α also induced IL-1β production at both mRNA and protein levels in the cells. IL-1β is expressed in an inactive precursor form that is cleaved by caspase-1 to generate mature IL-1β (Dinarello 1996). To determine the form of secreted IL-1β, the culture supernatants of MH7A stimulated with or without CSC for 24 h were analyzed by western blotting with an antibody against human IL-1β. Only precursor IL-1β was detected in the control cells. In contrast, both precursor and mature IL-1β were detected in the CSC-stimulated cells (Fig. 2C). CSC also augmented the caspase-1 activity in the cells stimulated with CSC for 24 h (Fig. 2D). As a sufficient number of primary cells were not available, we used MH7A in the subsequent experiments to investigate the mechanism of CSC on IL-1β mRNA induction. The cells were transfected with pGL4.10-IL-1β (−3791), which contained −3791 upstream of the transcriptional start of the human IL-1β gene, and treated with CSC for 24 h, and then luciferase activity was measured. As shown in Fig. 2E, CSC dose-dependently induced the promoter activity of the IL-1β gene in MH7A.

Effects of CSC on the expression of mRNA, protein, and gene activation of IL-1β in MH7A. Cells were treated with vehicles (control) and varying concentrations of CSC or TNF-α (5 U/mL) for 24 h.
Analysis of the IL-1β gene activation by CSC in MH7A
The mechanism of the IL-1β gene activation by CSC in MH7A was analyzed by using reporter plasmids with or without mutations. As shown in Fig. 3A, there are 4 NF-κB sites in the promoter region of the IL-1β gene. The far distal site was at −3771 to −3762 bp, the second one −3105 to −3096 bp, and the third one −2787 to −2778 bp, and the most proximal site was −308 to −249 bp. A series of 5′-deletion mutants of the IL-1β promoter genes was constructed to define the responsive region to CSC. After transfection with the plasmids, cells were treated with or without CSC for 24 h, and then the luciferase activity was measured. As shown in Fig. 3A, cells transfected with pGL4.10-IL-1β (−3791), pGL4.10-IL-1β (−3387), and pGL4.10-IL-1β (−2844) responded to CSC, and their response was increased according to the length of the promoter region. In contrast, pGL4.10-IL-1β (−2371) and pGL4.10-IL-1β (−474) did not respond to CSC, suggesting that 3 NF-κB sites present at the distal region were important for the responsiveness to CSC. To determine whether these 3 NF-κB sites are actually important and which of them is most critical to the responsiveness to CSC, a series of deletion mutant of the NF-κB sites was constructed, and measured the responsiveness to CSC. As shown in Fig. 3B, mutant 1 was most effective in the loss of responsiveness to CSC, then mutant 2, and mutant 3, indicated that each NF-κB site was important, and the far distal site was most important for the responsiveness to CSC.
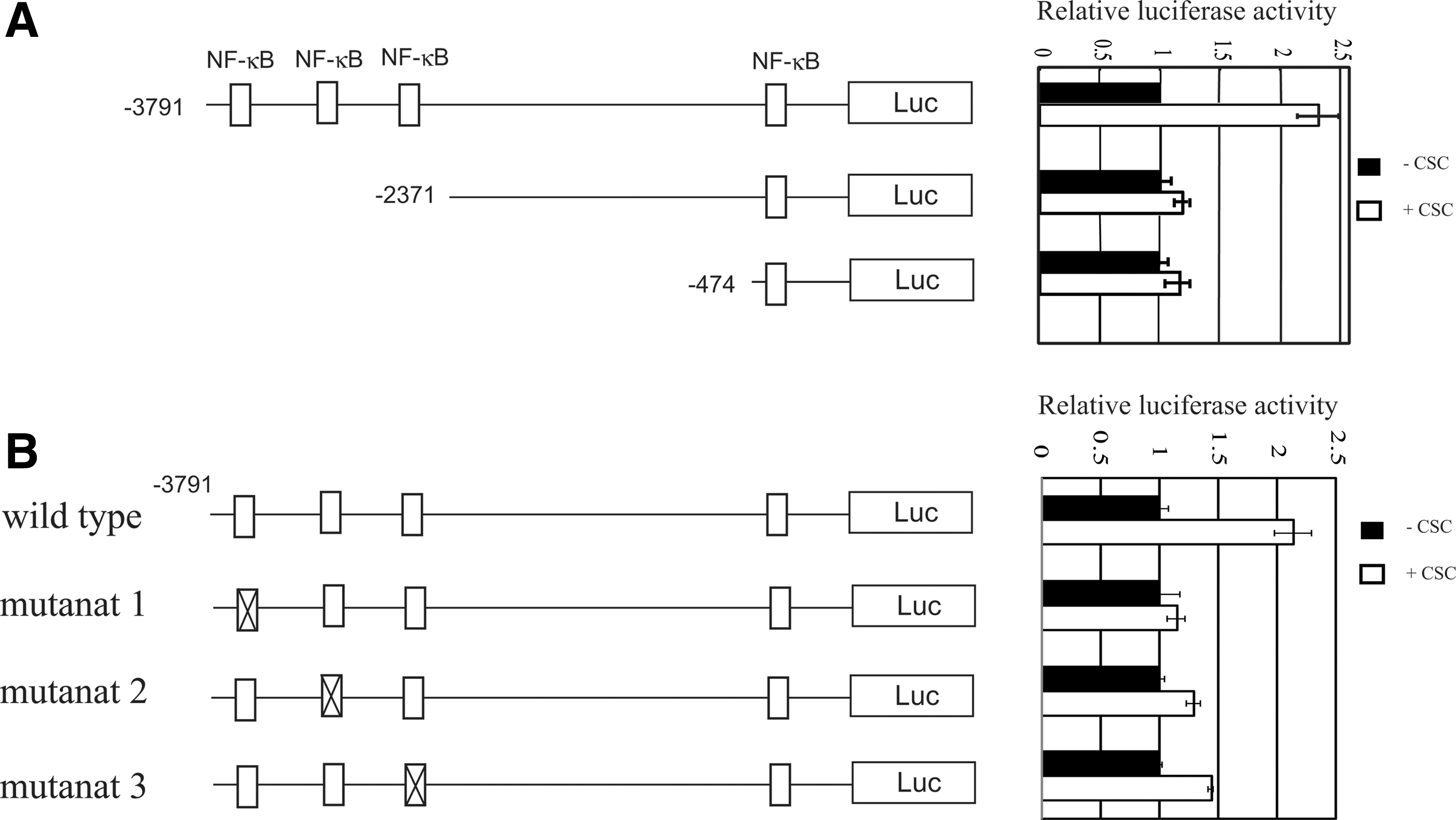
Deletion analysis of the 5′-flanking region of the human IL-1β gene in MH7A.
CSC-induced NF-κB activation is critical for the IL-1β mRNA induction by CSC in MH7A
To determine whether CSC activates NF-κB, MH7A was transfected with pGL4-(Ig-κB)6-Luc, which contains 6 tandem repeats of Ig-κB, and treated with or without CSC for 24 h, and then luciferase activity was measured. As shown in Fig. 4A, CSC dose-dependently increased the luciferase activity. NF-κB activation was confirmed by the CSC-induced downregulation of the IκB level, which began after 5 min of stimulation with CSC and continued up to 30 min (Fig. 4C), and translocation of p65 from the cytoplasm to the nuclei (Fig. 4E). We next examined whether the blocking of NF-κB activation leads to the inhibition of CSC-induced IL-1β mRNA expression. PDTC, an inhibitor of NF-κB, inhibited CSC-induced NF-κB-dependent luciferase activity (Fig. 4B), downregulation of IκB level (Fig. 4D), translocation of p65 from the cytoplasm to the nuclei (Fig. 4E), and IL-1β mRNA expression (Fig. 4F). These results indicate that CSC activated NF-κB, which leads to IL-1β mRNA induction.
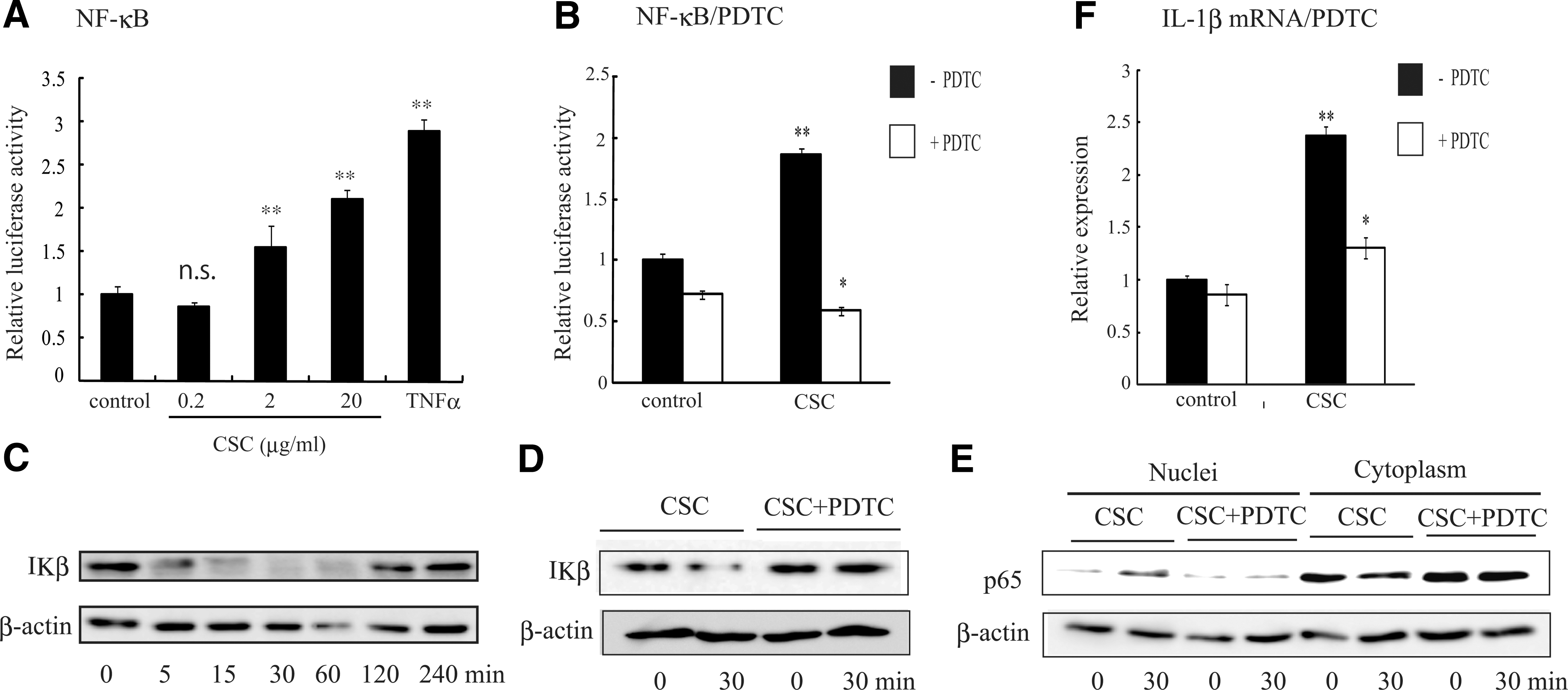
NF-κB activation by CSC. MH7A cells were transfected with pGL4-(Ig-κB)6-Luc and pGL4.74[hRlucTK], and treated with vehicles, varying concentrations of CSC or TNF-α (5 U/mL)
NF-κB binding to the promoter region of the IL-1β gene
The luciferase assay using the promoter region of the IL-1β gene indicated that 3 distal NF-κB sites, especially the far distal site, are important for the responsiveness to CSC (Fig. 3). To determine whether p65 was bound to the NF-κB site in response to CSC, the promoter pull-down assay was performed by using the wild-type promoter fragment covering from −3791 to −2711 bp and mutant fragments, which lack double or triple NF-κB sites as indicated in Fig. 5. The wild-type promoter bound p65 in response to CSC, but not to vehicle. The binding of p65 was mostly lost in the mutants that lack the far distal NF-κB site (mutants 5, 6, and 7). In contrast, the binding was slightly diminished by the loss of other NF-κB-binding sites (mutant 4). These findings indicate that p65 binding was parallel to the luciferase activity.

NF-κB binding to the promoter region of the IL-1β gene in response to CSC. Cells were treated with vehicles or CSC (20 μg/mL) for 30 min, and then nuclear extracts were obtained. The promoter pull-down assay was performed by using the indicated DNA fragments covering from −3791 to −2711 bp in the promoter region of the IL-1β gene. The hatched box indicates the mutation site. The level of DNA bound p65 was determined by western blotting with an antibody against p65. The p65 level in the nuclear extracts from the same number of cells was also indicated.
CSC induced NF-κB activation, IL-1β promoter activity, and IL-1β mRNA expression in a manner dependent on AhR
To determine whether the effect of CSC was mediated through AhR, the effect of α-NF, an antagonist for AhR, was determined on the CSC-induced NF-κB activation, IL-1β promoter activity, and IL-1β mRNA upregulation in MH7A. B[a]P, a PAH contained in CSC, was used as a reference stimulator. As shown in Fig. 6, B[a]P in a manner similar to CSC induced NF-κB activation, IL-1β promoter activity, and IL-1β mRNA upregulation in MH7A. α-NF at the dose that alone does not exhibit any effect completely inhibited all these effects of CSC and B[a]P (Fig. 6A–C). The mRNA of CYP1A1, a representative gene induced by PAHs (Watson and Hankinson 1992), was also induced by CSC and B[a]P, and was inhibited by α-NF (Fig. 6D). The inhibitory effect of α-NF on CSC-induced NF-κB activation was also confirmed by the reversal effects on the CSC-induced IκB degradation and p65 translocation from the cytoplasm to the nuclei (Fig. 6E, F). These results suggest that CSC-induced NF-κB activation, IL-1β promoter activity, and IL-1β mRNA upregulation are mediated through AhR.
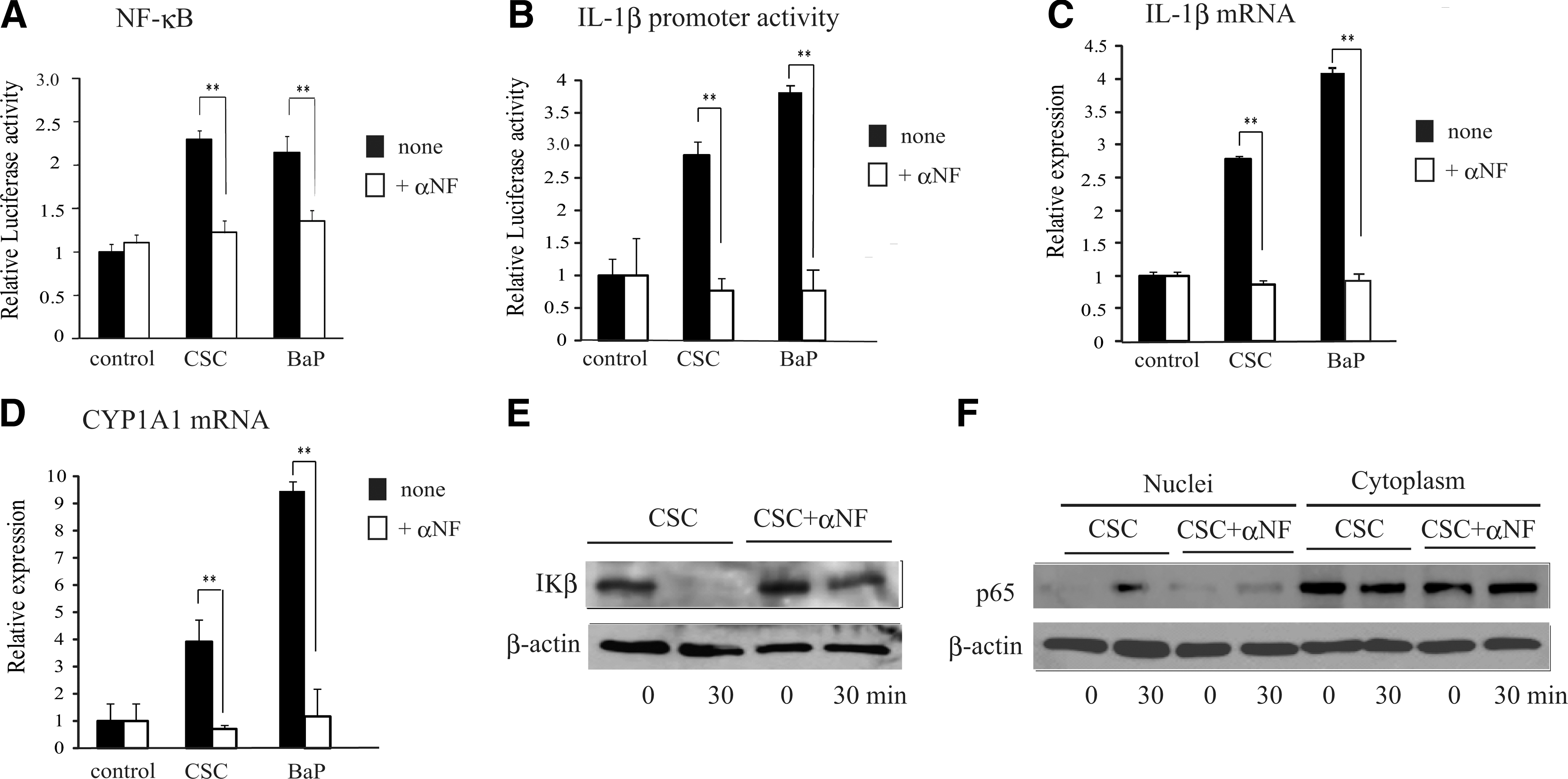
Effects of α-NF on the CSC-induced activation of the NF-κB and IL-1β promoter, and induction of IL-1β and CYP1A1 mRNA.
Discussion
In this study, we showed that mainstream CSC induced IL-1β mRNA from RA-patient derived synovial fibroblasts and RA-patient derived cell line MH7A, but not from OA patient-derived synovial fibroblasts. In parallel to the mRNA level, CSC induced IL-1β protein from MH7A. Interestingly, TNF-α also induced IL-1β mRNA from 2 of 3 RA-patient derived synovial cells and MH7A, but only slightly induced in 1 of 3 OA patient-derived synovial cells. Therefore, RA-patient derived synovial fibroblasts are sensitive not only to CSC but also to TNF-α, but OA patient-derived synovial cells were not responsive to these stimuli. RA synovial fibroblasts (RASF) are characterized by their activation condition, such as production of proinflammatory cytokines, proangiogenic factor production, including IL-8, vascular endothelial growth factor, basic fibroblast growth factor, and transforming growth factor-β, tumor-like transformation, enhanced aggressive and invasiveness, enhanced expression of adhesion molecules, MMPs, cell cycle-related gene expression, and high transcription of egr-1, fos, fun and myc, and activation of NF-κB (Huber and others 2006). Therefore, our findings support the concept that RASF are in a state of constitutive activation. In contrast, OA patient-derived synovial cells are static. IL-1β is expressed in an inactive precursor form that is cleaved by caspase-1 to generate mature IL-1β (Dinarello 1996). It is reported that RASF mostly produces precursor IL-1β (Kobayashi and others 1994). CSC induced both precursor IL-1β and mature IL-1β. CSC also augmented the activity of caspase-1.
Using MH7A, the mechanism of IL-1β mRNA induction by CSC was investigated, because a sufficient number of primary patient-derived cells were not available to perform the subsequent experiments. CSC dose-dependently induced the promoter activity of the IL-1β gene in MH7A, indicating that CSC induced IL-1β mRNA at the transcriptional level. Studies on the molecular analyses of the IL-1β gene revealed that the region between −2982 and −2795 bp upstream from the transcription start site was necessary for phorbol 12-myristate 13-acetate (PMA) induction of IL-1β in human monocytic THP-1 and U937 cells (Bensi and others 1990); C/EBPβ/NF-IL-6 site between −2894 and −2864 bp and NF-β1 site between −2850 and −2831 bp were for LPS induction in THP-1 and fibroblast cells (Shirakawa and others 1993); CRE-like site between −2761 and −2753 bp was for LPS, PMA, or cAMP induction in THP-1 and U937 cells (Gray and others 1993); the AP-1 site between −2982 and −2748 bp was for PMA induction in THP-1 cells (Serkkola and Hurme 1993); NF-IL-6 sites between −90 and −82 bp and between −40 and −32 bp were for LAM, LPS, or TNF-α induction in THP-1 cells (Zhang and Rom 1993); NF-κB sites between −296 and −286 bp was for PMA, Sendai virus, IL-1β, LPS, PMA, or cAMP induction in U937 cells (Hiscott and others 1993, Cogswell and others 1994). Cogswell also found the NF-κB site between −2800 to −2720 bp; however, the mutation of this site did not affect the gene activation of IL-1β by LPS, PMA, and dibutyryl cyclic AMP (Cogswell and others 1994). We analyzed which region of the promoter is important for the CSC-induced IL-1β gene activation by using a series of deletion mutants of the IL-1β reporter gene and deletion mutants in the NF-κB sites. The NF-κB site between −296 and −286 bp was not important for the CSC-induced promoter activation, probably due to the different experimental conditions. We found that each distal 3 NF-κB sites at −3771 to −3762 bp, −3105 to −3096 bp, and −2787 to −2778 bp was critical. Therefore, our findings revealed 3 novel NF-κB sites as critical for the gene activation of IL-1β by CSC. Among the 3 NF-κB sites, the far distal site between −3771 and −3762 bp was most important for the responsiveness to CSC, which was confirmed by p65 binding to the DNA fragments in response to CSC.
We found that CSC activated NF-κB in MH7A by the reporter assay using pGL4-(Ig-κB)6-Luc, which was confirmed by the downregulation of IκB and translocation of p65 from the cytoplasm to nuclei. We also found that PDTC, an inhibitor of NF-κB, inhibited both the CSC-induced NF-κB activation and IL-1β mRNA expression. In accord with our results, it is reported that CSC induced NF-κB activation in several human cell lines, U937, T-cells (Jurkat), and lung carcinoma cell line (H1299) (Anto and others 2002). It is of note that endotoxin was undetectable in our CSC preparation, and we added polymyxin B to the assay to rule-out the effect of the undetectable level of endotoxin. These results indicate that CSC, other than endotoxin, activates NF-κB, which leads to the IL-1β mRNA induction by upregulating promoter activity in MH7A.
We have reported that PAHs, such as 3-MC, B[a]P and TCDD, induced IL-1β mRNA in MH7A. We also reported that CSC induced IL-1α, IL-1β, IL-6, and IL-8 at both mRNA and protein levels in the cells (Shizu and others 2008). More than 4,000 chemicals are estimated to be present in CSC, and the amount of total mainstream smoke PAHs is ranging from 1 to 1.6 μg per cigarette (Ding and others 2005). Therefore, we expected that PAHs may contribute to the effect of CSC. In this study, an antagonist for AhR, α-NF, completely inhibited the CSC-induced NF-κB activation, IL-1β promoter activity, and IL-1β mRNA upregulation in MH7A. Therefore, the effects of CSC appeared to be mediated through AhR. It is reported that AhR physically interacts with NF-κB, and induces activation or inhibition of NF-κB activity not only by classical (canonical) pathway, but also by an alternative pathway through forming AhR/RelA (p65) and AhR/RelB heterodimers, respectively (Tian 2009, Vogel and Matsumura 2009). The differential consequences of the AhR/NF-κB interaction may be due to the difference of the cell type or AhR agonist.
The nonresponsiveness of OA patient-derived synoviocytes to CSC may be due to the expression level of AhR. Kobayashi reported that higher AhR mRNA and protein levels were observed in RA synovial tissue than in OA tissue, and in accord with our previous report and present findings, TCDD increased IL-1β mRNA levels via AhR in RA synoviocytes (Kobayashi and others 2008). The amount of PAHs in the synovial fluid is not known. However, the dose of 2 μg/mL CSC, which is able to induce IL-1β mRNA in RASF, is reachable if an individual with 60-kg body weight takes only 13 cigarettes, in assuming that all the smokes were adsorbed and distributed equally in the body. Among the chemicals in CSC, PAHs and other hydrophobic chemicals are readily absorbed into adipose tissue and can be accumulated for a long period of time. Actually, heavy smokers daily intake much more cigarettes, and the number reaches up to an uncountable level for a long period of time. Supporting this issue, a recent report indicates that AhR activation, indicated by AhR repressor and CYP1A1 gene expression, was found only in synovia from RA patients who smoked (Kazantseva 2012). It is also of note that we used mainstream CSC that has passed through the cigarette filter. Therefore, even if cigarette filter reduces the amount of smoke, tar, and fine particles, the filter-passed smoke contains hazardous chemicals. We have reported that CSC, either mainstream or sidestream, augmented the induction and clinical development of arthritis in collagen-induced arthritis (CIA) (Chujo and others 2010, Okamoto and others 2011). The positive involvement of AhR was reported in CIA using an AhR-deficient mouse (Nakahama and others 2011). Collectively, our findings support that exposure to cigarette smoke exacerbates the development of RA and support the etiological role of cigarette smoking in RA (Onozaki 2009). Indeed, brief exposure to cigarette smoke increases the serum level of proinflammatory cytokines for as long as 3 h (Flouris and others 2009) and induces AhR activation in vivo (Kasai and others 2006).
Footnotes
Acknowledgments
We thank Dr. Yuka Itoh for her help. This work was supported in part by a grant from the Smoking Research Foundation.
Author Disclosure Statement
No competing financial interests exist.