
Abstract
After several decades of intense clinical research, the great promise of Type I interferons (IFN1) as the anticancer wonder drugs that could cure or, at the very least, curb the progression of various oncological diseases has regrettably failed to deliver. Severe side effects and low efficacy of IFN1-based pharmaceutics greatly limited use of these drugs and further reduced the enthusiasm of clinical oncologists for future optimization of IFN1-based therapeutic modalities. Incredibly, extensive clinical studies to assess the efficacy of IFN1 alone or in combination with other anticancer drugs have not been paralleled by an equal scope in defining the determinants that confer cell sensitivity or refractoriness to IFN1. Given that all effects of IFN1 on malignant and benign cells alike are mediated by its receptor, the mechanisms regulating these receptor cell surface levels should play a paramount role in shaping the magnitude and duration of IFN1-elicited effects. These mechanisms and their role in controlling IFN1 responses, as well as an ability of a growing tumor to commandeer these events, are the focus of our review. We postulate that activation of numerous signaling pathways leading to elimination of IFN1 receptor occurs in cancer cells and benign cells that contribute to tumor tissue. We further hypothesize that activation of these eliminative pathways enables the escape from IFN1-driven suppression of tumorigenesis and elicits the primary refractoriness of tumor to the pharmaceutical IFN1.
Had Enough of Interferon?
T
However, as often experienced throughout a century-long history of cancer treatment, the initial euphoria regarding a given strategy is gradually substituted with a more cautious approach. Intrinsic to molecularly targeted therapeutics is the rapid development of resistance due to target-specific or pathway-specific acquired mutations (Barouch-Bentov and Sauer 2011; Watzka and others 2011). This has prompted modern oncopharmacology to revisit its strategy and seek a greater potency at the expense of specificity [reviewed in Fojo (2008)]. Among agents that can combat cancer growth and progression through pleiotropic mechanisms are antitumorigenic cytokines, the interferons (IFNs). While 3 diverse types of IFN are known (de Weerd and Nguyen 2012; George and others 2012), most pharmaceutically produced IFNs (including IFN-α and IFN-β) belong to Type I interferon (IFN1). Driven perhaps by frustration with targeted therapies, there is renewed interest in using IFN1 as an anticancer drug (Kujawski and Talpaz 2007; Hasselbalch 2011). Accordingly, we will focus on IFN1 in this review.
IFN1 acts on cells by engaging a cell surface-localized cognate IFN1 receptor. Formation of the ligand–receptor complex triggers the activation of JAK-STAT and other signaling pathways described in many excellent reviews [see Aaronson and Horvath (2002), Platanias (2005), and reviews in this issue]. This signal transduction leads to the activation of diverse genes that mediate the direct and indirect effects of IFN1 on tumor growth and progression and on antitumor immunity. As outlined in other reviews included in the issue of this journal and elsewhere (Gresser 2007; Bekisz and others 2010), the effects of IFN1 on tumors are very potent, yet extremely pleiotropic and still poorly understood.
This paucity of knowledge on the mechanisms by which IFN1 works is perplexing given that the history of IFN1 use against various oncological diseases is almost 40 years old. Despite the known experimental potency of IFN and the extensive inclusion of IFN1 alone and in combination with other drugs into therapeutic modalities against these diseases, the enthusiasm for the continuous use of IFN1 is undermined by an unsatisfactory ratio between clinical efficacy and the severity of its side effects [reviewed in Bracarda and others (2010)]. Additional drawbacks include IFN1's relatively high cost of production (of recombinant protein versus a small molecule), issues with its bioavailability [that could be compensated by pegylation at the expense of efficacy (George and others 2012)], and as seen in patients with multiple sclerosis, its eventual inactivation through the development of neutralizing antibodies (Strayer and Carter 2012). All these factors aggravate the ongoing doubts of clinicians as to whether IFN1 use has any merit in the management of oncologic patients.
Should oncologists who put in a tremendous effort to determine the practicality of optimizing IFN1 and its combinations regimens against diverse cancers [reviewed in Kirkwood and Ernstoff (1984)] quit using IFN as an anticancer agent altogether? One needs to understand the mechanisms that control the sensitivity of cancers to IFN1 prior to answering this question. After all, despite initial disappointment with tamoxifen efficacy against all breast cancers, the mechanistic studies that led to the molecular understanding of breast cancer types led to the restriction the use of tamoxifen on estrogen receptor-positive patients (Ciocca and Elledge 2000). Similarly, additional molecular studies identified of a group of patients whose breast cancers overexpressed Her2 and are sensitive to Herceptin (Ahmed and others 2011). Other groups of patients with breast cancer who lack the molecular targets for tamoxifen or Herceptin do not benefit from these treatments.
Accordingly, as all effects of IFN1 are mediated by its binding to the IFN1 receptor on the cell's surface, one could anticipate the futility of using IFN1-based drugs to treat patients whose cells do not present this receptor. The goal of this review was to outline the mechanisms regulating the cell surface expression of the IFN1 receptor and to analyze how these mechanisms regulate the sensitivity of malignant and benign cells that form tumor tissue to IFN1. Our future ability to modulate these parameters toward the reactivation of antitumorigenic effects of either endogenous or pharmaceutical IFN1 will be discussed.
Cell Surface Density of Cognate Receptors Determines the Extent of Cellular Responses to IFN1
The essential role of the IFN1 receptor in the regulation of cellular responses to IFN1 has been demonstrated by numerous studies and has been extensively reviewed elsewhere (Uze and others 2007). A functional receptor that belongs to the class II helical cytokine receptor family is formed by 2 subunits, IFNAR1 and IFNAR2. The extracellular domain of the latter chain contains characteristic fibronectin III-like domains and mostly confers affinity to IFN1. Affinity of the extracellular domain of IFNAR1 varies toward diverse subtypes of IFN1 and correlates specifically to the intrinsic ability of these subtypes to elicit an antiproliferative effect (Jaitin and others 2006; Jaks and others 2007; Kalie and others 2007). Recent advances in solving the structure of the ternary signaling complexes between these subunits and ligand (Thomas and others 2011) have solidified the current paradigm that the dynamics and the stability of the ligand–receptor complex are critical regulators of specific intracellular cascades and the ensuing cellular responses elicited by diverse subtypes of IFN1 (Kalie and others 2008).
While studies in IFNAR1 knockout mice left no doubt about the essential role of the receptor in IFN1 responses (Muller and others 1994; Hwang and others 1995), there have been certain arguments as per how important cell surface receptor density is in determining the extent of cellular responses. These doubts stem from early ligand binding studies that suggested that even partial receptor occupancy might suffice for mounting a robust biological effect (Pfeffer and Donner 1990), suggesting that intermediate changes in the receptor cell surface levels may have very subtle effects on the extent of IFN responses. However, this point of view is not supported by genetic evidence. While IFNAR1-null mice are utterly unresponsive to IFN1, the heterozygous mice harboring just one copy of the Ifnar1 gene exhibit an intermediate phenotype (Hwang and others 1995), suggesting that quantitative changes in IFNAR1 levels are pivotal for fine-tuning the magnitude and duration of cellular responses to IFN1.
Furthermore, it has been noted that the antitumorigenic effects of IFN1 might be especially sensitive to cell surface receptor levels and their ability to interact with greater doses of these cytokines. While the antiviral effects of IFN1 could be efficiently achieved in cells that express a limited number of receptors, a high cell surface receptor density and maximal receptor occupancy by relatively high doses of ligands are required to mount an efficient antiproliferative effect (Moraga and others 2009; Levin and others 2011). Accordingly, Schreiber and others have proposed that responses to IFN could be classified as robust (such as antiviral effects) or tunable (such as antiproliferative or proinflammatory) (Levin and others 2011).
The diverse consequences of decreasing the receptor levels between these scenarios for robust responses, elicited by low levels of ligand engaging a few receptors, and tunable responses, requiring high receptor density are illustrated in Fig. 1. What has to be emphasized at this point is that substantial levels of receptors on the cell surface are essential for eliciting antitumorigenic effects (Moraga and others 2009; Levin and others 2011). Indeed, the expression of the IFN1 receptor correlated with IFN-α-mediated growth arrest (Eguchi and others 2000) and apoptosis in the tumor samples (Mejia and others 1999; Vitale and others 2007). Furthermore, expression of either IFNAR1 (Colamonici and others 1994) or IFNAR2 (Wagner and others 2004) in diverse cancer cell lines led to decreased proliferation and viability and stimulated differentiation.
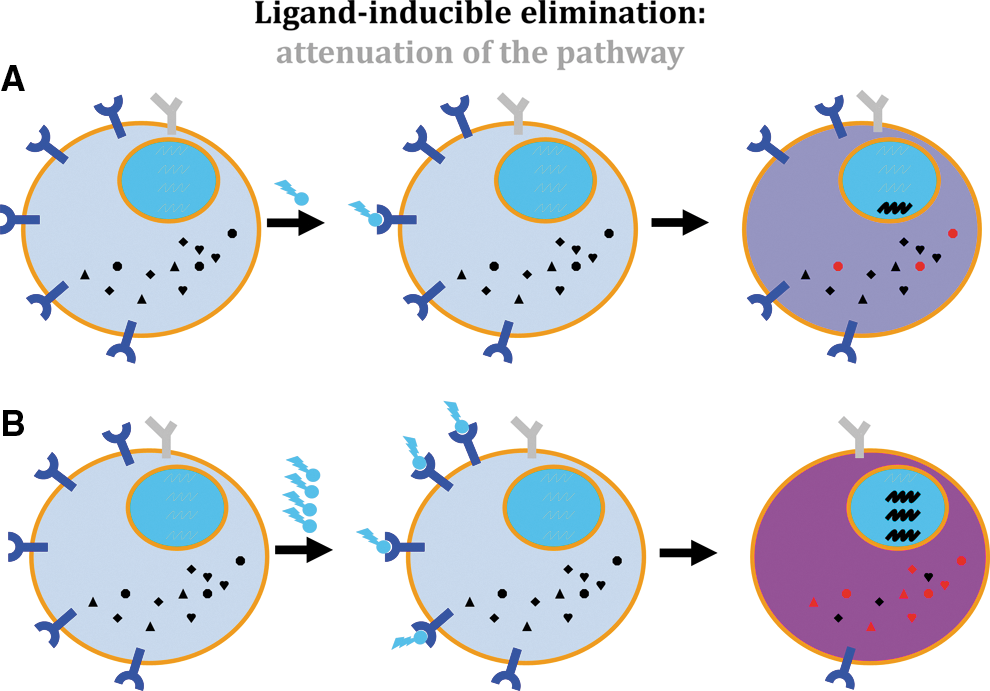
Ligand-inducible elimination.
In all, the levels of receptor on the cell's surface determine the sensitivity of cells to endogenous and pharmacologic IFN and control the magnitude and duration of antitumorigenic effects elicited by these cytokines. How are these levels of receptor regulated?
Regulation of IFN1 Receptor Cell Surface Levels
IFNAR1 and IFNAR2 mRNAs are constitutively expressed in multiple cell types (Hensley and others 1993). Among the numerous mechanisms that constitute a negative feedback tempering the effects of IFN1, downregulation of a receptor is considered to be the most specific and rapid regulation (Coccia and others 2006). Experiments with radiolabeled ligands showed that IFN-α treatment downregulates IFN-α/β receptor levels on the surface of cultured cells (Branca and others 1983) and in cells from patients treated with IFN-α (Maxwell and others 1985; Billard and others 1986).
Rapid endocytosis and subsequent lysosomal degradation represent a major pathway for regulating functional levels of the IFN1 receptor (Kumar and others 2003; Uze and others 2007). This pathway constitutes an important and specific mechanism for restricting the extent and duration of IFN-α signaling (Stark and others 1998; Coccia and others 2006). Endocytosed IFNAR2 demonstrates a propensity to recycle back to the cell surface (Marijanovic and others 2007), whereas endocytosis of IFNAR1 usually leads to its degradation [reviewed in Huangfu and Fuchs (2010) and Fuchs (2012)]. As endocytosis of the entire receptor is often determined by an assortment of mechanisms accelerating endocytosis and degradation of IFNAR1 (Kumar and others 2003; Uze and others 2007), most of this review will be dedicated to discussing these mechanisms.
The IFNAR1 protein is rapidly downregulated and degraded upon internalization in response to IFN-α (Constantinescu and others 1994, 1995). Internalization of IFNAR1 occurs via the clathrin-dependent pathway that relies on the recognition of an exposed endocytic motif by the AP2 endocytic machinery complex (Kumar and others 2007a, 2008). In human IFNAR1, this linear motif is tyrosine based (YXXΦ linear endocytic motif, where Φ is a hydrophobic amino acid residue). In unstimulated cells, this motif is masked by associated TYK2 kinase (Kumar and others 2008). As a result, human cells lacking TYK2 are characterized by a high rate of constitutive endocytosis and degradation of IFNAR1 (Ragimbeau and others 2003); these cells exhibit low levels of IFNAR1 (Gauzzi and others 1997). A single example of human melanoma cells that express neither TYK2 nor IFNAR1 has been reported (Pansky and others 2000).
Intriguingly, phosphorylation of the tyrosine residue within the YXXΦ linear endocytic motif (that is likely to occur in IFN1-treated cells) is expected to decrease the affinity of this motif to binding with AP2 (Bonifacino and Traub 2003). Accordingly, the fact that IFN1 still promotes IFNAR1 internalization suggests an important role for a protein tyrosine phosphatase that could counteract this phosphorylation and enable ligand-induced receptor downregulation. Indeed, our recent studies identified PTP1B phosphatase as a key regulator of IFNAR1 endocytosis, stability, and signaling (Carbone and others 2012).
The tyrosine-based motif-dependent regulation of IFNAR1 endocytosis and stability is likely to occur in a majority of vertebrate species that harbor IFN1 signaling. A notable exception from this rule is mouse IFNAR1 that does not contain a conserved YXXΦ sequence, and whose internalization and ensuing degradation likely rely on other types of endocytic motifs (possibly di-leucine/isoleucine repeats). Accordingly, cells from mice lacking TYK2 display normal levels of cell surface IFNAR1 (Karaghiosoff and others 2000; Shimoda and others 2000), and cells from mice lacking PTP1B are not defective in IFNAR1 internalization (Carbone and others 2012).
Regardless of the motif type required for receptor internalization, the latter process (in human and mouse cells alike) is robustly augmented by IFN1-induced ubiquitination of IFNAR1 (Kumar and others 2003, 2007a). This ubiquitination occurs on many ubiquitin acceptors within the cytoplasmic tail of IFNAR1. However, the polyubiquitination of a specific cluster of lysine residues (such as K501, 525, and 526 within human IFNAR1) appears to be critical (Kumar and others 2004, 2007a; Fuchs 2012). This ubiquitin transfer to either K48-linked or K63-linked polyubiquitin chains on these lysines is likely to involve 2 diverse E2 ubiquitin-conjugating enzymes: Cdc34 and Ubc13/Uev1 (our unpublished data). Both types of linkages contribute to the maximal rate of IFNAR1 internalization. Furthermore, overall ubiquitination plays a key role in the postinternalization trafficking of IFNAR1-containing vesicles toward late-endosomal and lysosomal intracellular compartments, where these vesicles undergo irreversible proteolysis (Kumar and others 2007a; Fuchs, 2012). This process is expected to be counteracted by specific deubiquitinating enzymes, whose nature and mode of function will be determined by future studies.
All types of ubiquitination of IFNAR1 are facilitated by the SCF-βTrcp2 E3 ubiquitin ligase (Kumar and others 2003). This is a Cullin-based ligase that combines an ubiquitin-ligating module (consisting of the C-terminal portion of Cullin1 and the associated ROC1/Rbx1 RING-domain protein; both contribute to the recruitment of the E2-ubiquitin conjugate), a tethering module (the N-terminal portion of Cullin1 and the associated Skp1), and a substrate recognizing subunit, the F-box protein βTrcp2 [also termed HOS, reviewed in Fuchs and others (2004)].
IFN1 induces IFNAR1 ubiquitination along with subsequent endocytosis and degradation by stimulating the recruitment of βTrcp2 to IFNAR1 (Kumar and others 2003, 2007a). Importantly, βTrcp2 interacts poorly (if at all) with unmodified IFNAR1, yet it is robustly recruited to this receptor upon its serine phosphorylation stimulated by IFN1 (Kumar and others 2003, 2004). These specific serine residues form a definitive phospho-degron motif DSGNYS that is conserved throughout IFNAR1 in all known species (for example, Ser535 and 539 in human IFNAR1, or Ser526 and 530 in mouse IFNAR1). Based on numerous structural and biochemical studies carried out on SCF-βTrcp E3 ubiquitin ligase and its model substrates (reviewed in Harper and Schulman (2006)], it has been predicted that the recruitment of βTrcp2 to the phospho-degron enables the juxtaposition of the ligase-bound E2–ubiquitin conjugate against the ubiquitin-acceptor sites (such as K501, 525, and 526 within human IFNAR1) and enables the transfer of ubiquitin to these sites or existing ubiquitin chains originated at these sites (Kumar and others 2004, 2007a).
As the IFN1-stimulated recruitment of βTrcp2 (and subsequent ubiquitination, endocytosis, and lysosomal degradation of IFNAR1 and ensuing desensitization of cells to the ligand) relies on the phosphorylation of phospho-degron serine residues, this phosphorylation is the most important, specific, and rate-limiting event in regulating the ability of cells to respond to IFN1. Signaling pathways leading to this phosphorylation and receptor elimination [termed eliminative signaling (Huangfu and Fuchs 2010)] and activation of this elimination in cancers are outlined below.
Eliminative Signaling Leading to IFNAR1 Degradation
Extracellular ligands (in our case, IFN1) interact with cognate receptors (IFNAR) to induce signaling cascades that result in a shift of the transcriptional program of a cell. The common proximal stem steps are followed by bifurcation into forward signaling leading to transcriptional changes and eliminative signaling. The latter accelerates the downregulation of an already-activated receptor and eventually terminates the signal origination (Fig. 2). Eliminative signaling toward IFNAR1 can be ligand inducible or ligand independent.
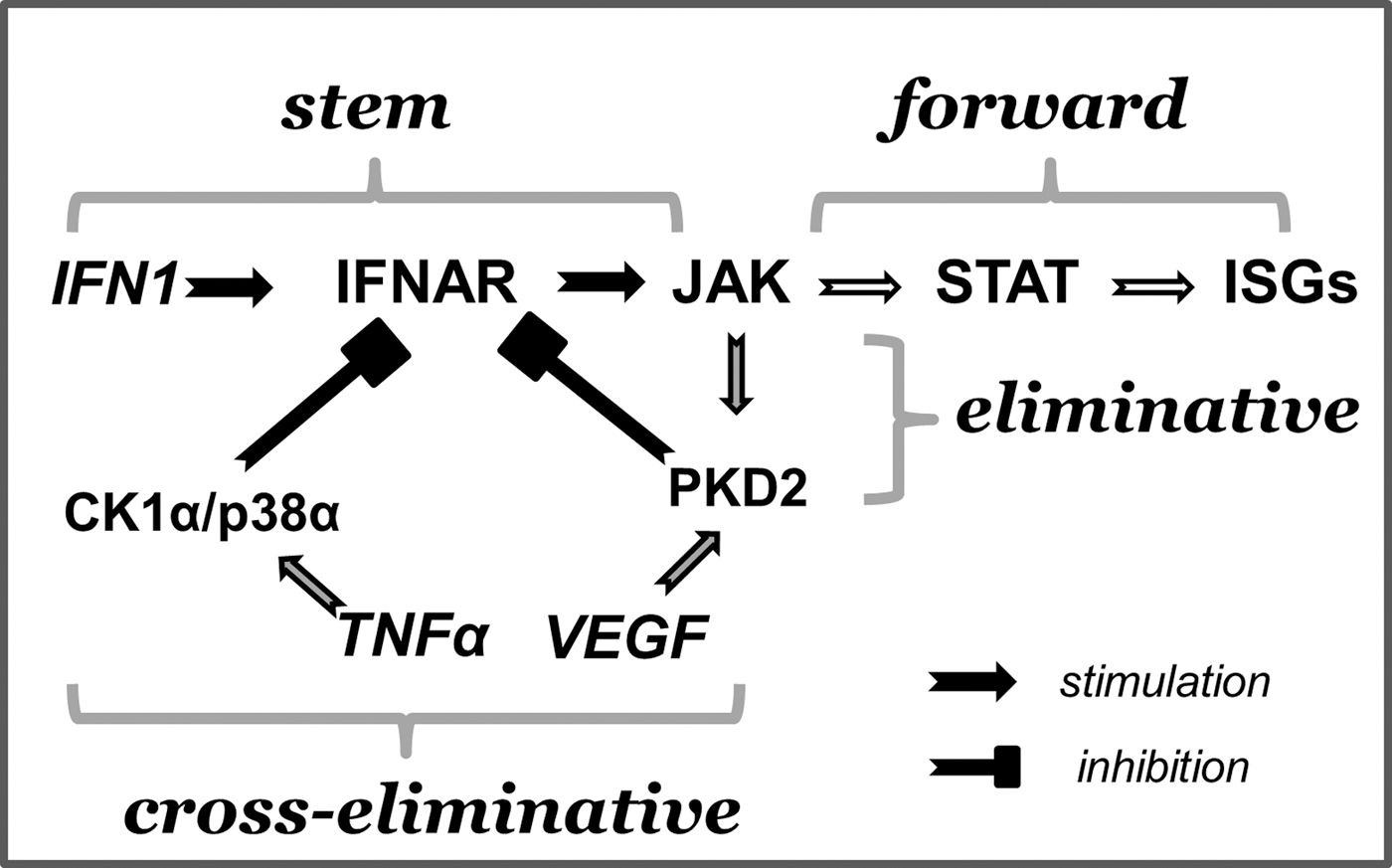
Eliminative signaling by IFN1. The concept of eliminative signaling (Huangfu and Fuchs 2010) is illustrated specifically on the example of IFN1 responses. IFN1 binds to the receptor (IFNAR) to activate JAK (stem signaling). JAK serves as a central signaling dispatcher that further distributes the cascades between forward signaling (ie, activation of STAT resulting in the transcriptional activation of ISGs) and eliminative signaling [ie, activation of protein kinase D2 (PKD2) and subsequent downregulation of the receptor]. Unrelated ligands such as vascular endothelial growth factor (VEGF) or tumor necrosis factor-α (TNF-α) could utilize PKD2 or basal pathway to eliminate the receptor without benefits of forward signaling and ISG induction.
The ligand-inducible signaling occurs within a canonical signaling cascade triggered by IFN1. This pathway has been delineated only recently despite numerous prior studies that demonstrated a decrease in cell sensitivity and ligand occupancy sites in cells already pretreated with IFN1 (Branca and others 1983; Maxwell and others 1985; Billard and others 1986). Early mechanistic clues to ligand-inducible elimination were provided by reports that described a negative regulatory domain within the distal part of the cytoplasmic tail of IFNAR1. The deletion of this domain prevented the ligand-inducible downregulation of the receptor and augmented cellular responses to IFN1 (Gibbs and others 1996; Basu and others 1998). The identification of a putative phospho-degron motif for the recruitment of βTrcp2/HOS within this domain led to the characterization of this E3 ubiquitin ligase as a major regulator of IFNAR1 ubiquitination and stability (Kumar and others 2003). It also resulted in the subsequent characterization of the phosphorylation of specific serines within this motif as a crucial event in receptor downregulation (Kumar and others 2004). As phospho-specific antibodies raised against these serines predominantly reacted with modified Ser535 in human IFNAR1 or with Ser526 in a mouse receptor (Kumar and others 2004), subsequent studies were focused on the protein kinases mediating phosphorylation of these specific residues. Accordingly, a hypothetical role of additional IFN1-inducible protein kinases that might target the secondary phosphoacceptors (Ser539 and Ser530) in receptor downregulation cannot be ruled out.
How does IFN1 stimulate the phosphorylation of IFNAR1 degron? A logical hypothesis would be that IFN1 induces a kinase that leads to this phosphorylation. Experiments using isogenic cell lines either lacking TYK2 or reconstituted with catalytically inactive TYK2 kinase (Marijanovic and others 2006) and pharmacologic inhibitors (Liu and others 2008) have demonstrated the role of JAK activity in this phosphorylation in IFN1-treated cells. Given that JAK do not efficiently phosphorylate serines (Aaronson and Horvath 2002; Seavey and Dobrzanski 2012), a search for an IFN1-inducible and JAK-dependent serine kinase continued for a few years until a combined approach of partial protein purification and inhibitory analysis identified protein kinase D2 (PKD2) as an essential ligand-stimulated Ser535/526 kinase (Zheng and others 2011c). PKD2 directly regulates the phosphorylation of IFNAR1 degron in IFN1-treated cells. This kinase also controls the IFN1-stimulated interaction between IFNAR1 and βTrcp2, the extent of the ubiquitination of IFNAR1, and the endocytic rate of this receptor and its half-life. Furthermore, PKD2 plays an important role in regulating the magnitude and duration of IFN1 signaling and its ability to mount an antiviral response (Zheng and others 2011c).
IFN1 treatment increases the ability of PKD2 to phosphorylate IFNAR1 degron motif through at least 2 independent mechanisms. First, JAK-mediated phosphorylation of PKD2 on Tyr438 within its plekstrin homology domain relieves the autoinhibitory conformation of PKD2 leading to the increased catalytic activity of this kinase (Zheng and others 2011a). This phosphorylation of PKD2 on Tyr438 is essential for IFN1-stimulated IFNAR1 ubiquitination, degradation, and restriction of its ability to signal downstream. Second, the recruitment of PKD2 to IFNAR1 is increased in cells treated with IFN1 in a manner that is poorly understood. However, the recruitment does not require the activation of TYK2 (Zheng and others 2011a), but may depend on some PKC species (our unpublished data) that are known to be induced by IFN1 (Redig and Platanias 2007).
The ligand-independent signaling (Fig. 3) that leads to IFNAR1 elimination may include cascades initiated by other stimuli through the activation of either PKD2 [eg, vascular endothelial growth factor, VEGF (Zheng and others 2011b)], or other kinases that utilize the basal pathway of IFNAR1 degradation (described in details below). Briefly, IFNAR1 degron undergoes weak constitutive phosphorylation by casein kinase 1α [CK1α (Liu and others 2009a)]. This process could be dramatically augmented by the phosphorylation of IFNAR1 on a priming site [Ser532 in human IFNAR1 or in Ser523 in a mouse receptor (Bhattacharya and others 2010)]. This priming phosphorylation is mediated by p38α stress-activated protein kinase (Bhattacharya and others 2011a) and can be triggered by an unfolded protein response [UPR (Liu and others 2009b)] or by inflammatory cytokines such as tumor necrosis factor-α [TNF-α (Huangfu and others 2012)].
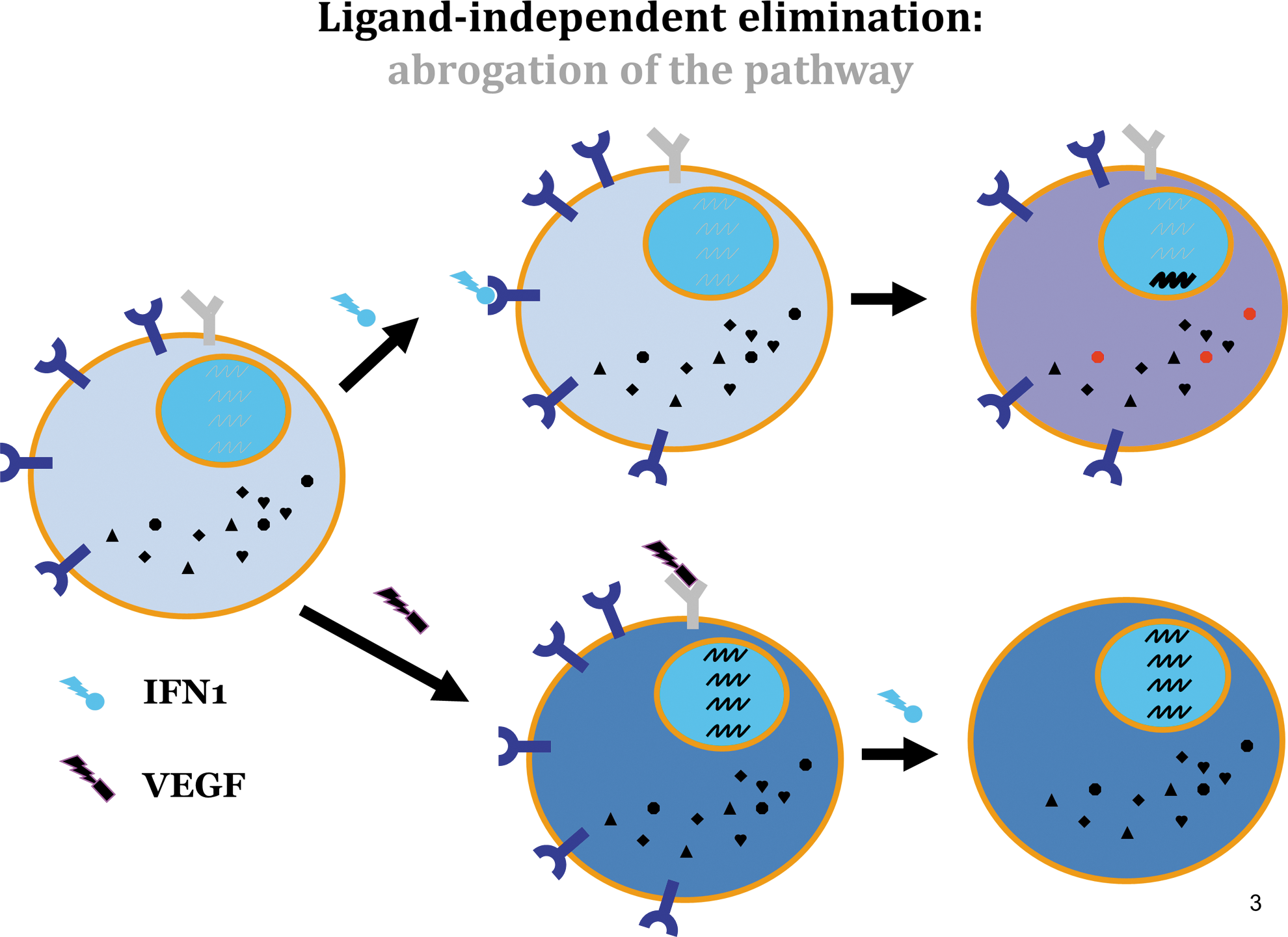
Receptor downregulation-inducing pathways that do not involve the exposure of cells to ligand (IFN1) such as VEGF→VEGFR→PKD2→pSer535 or TNF-α→TNFR→p38α→pSer532→CK1α→pSer535 are collectively termed cross-eliminative signaling. While all eliminative pathways leading to the downregulation of IFNAR1 invariably converge at the phosphorylation of Ser535/526 within the degron motif (Fig. 4), the functional consequences of the ligand-inducible elimination (Fig. 1) could dramatically differ from those triggered by cross-eliminative signals (Fig. 3). Cells that are first exposed to IFN1 execute a complete signaling program that includes forward signaling, which culminates in the transcription/translation of the IFN-stimulated genes and the execution of their direct and indirect antitumorigenic effects. In these cells, ligand-inducible phosphorylation and downregulation of IFNAR1 merely reduce the sensitivity of an activated cell to its next exposure to a ligand (Figs. 1 and 3).
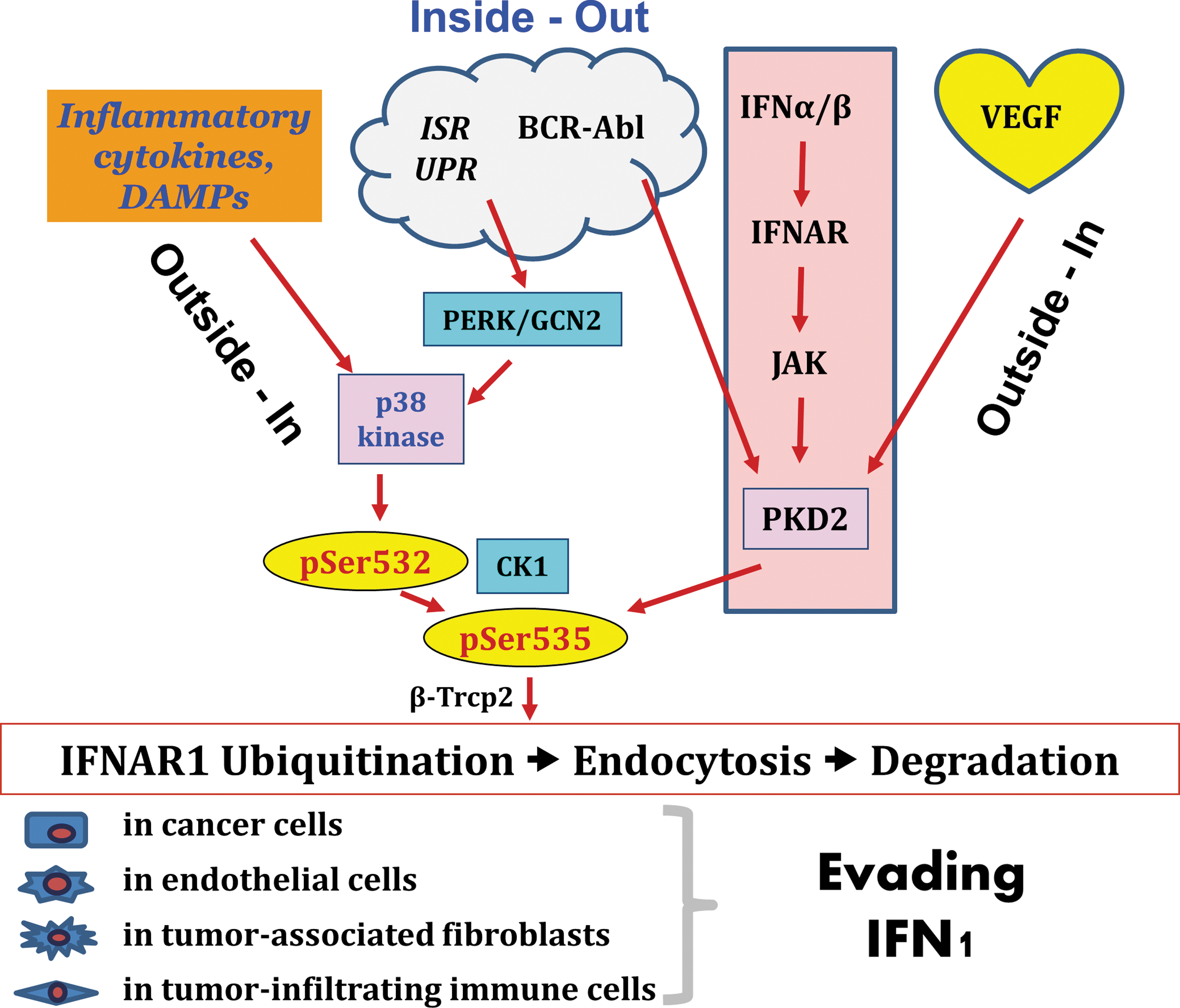
Sources and targets of eliminative signaling. Examples of stimuli that activate inside-out or/and outside-in modes of eliminative signaling leading to acceleration of IFNAR1 degradation and desensitization of cells to antitumorigenic effects of IFN1 are depicted. These stimuli include intracellular (for example, integrative stress response, ISR) and extracellular (eg, danger-associated molecular patterns, DAMPs) triggers. As a result of these eliminative pathways, levels of IFN1 receptor and sensitivity to IFN1 will be decreased in cells that contribute to the tumor formation, including cancer cells themselves as well as benign cells of stromal, angiogenic, and immune compartments leading to an overall effect of attenuating direct and indirect antitumorigenic effects of endogenous or pharmacologic IFN1. Color images available online at
Conversely, cells that are first exposed to a cross-eliminative stimulus (eg, VEGF or TNF-α) are expected to decrease the surface levels of IFNAR1 without having the benefit of any induction of IFN-stimulated genes (Fig. 3). Moreover (for the period of time required for IFNAR1 resynthesis, maturation, and trafficking to the plasma membrane), these cells will be rendered insensitive to any amount of endogenous or pharmacologic IFN1. In the next section of this review, we will examine the hypothesis that cross-eliminative signaling represents a central mechanism by which cellular compartments of tumor tissue are deprived from the ability to respond to future encounters with IFN1 and can consequently evade its antitumorigenic effects.
PKD2-dependent IFNAR1 elimination in tumors
Within the context of IFN-elicited responses, the antitumorigenic effect of activated IFN-stimulated genes coexists with PKD2-dependent downregulation of IFNAR1. However, aberrant activation of PKD2 in tumor cells may pre-emptively eliminate the receptor, rendering these cells refractory to subsequent IFN1 exposure. It is plausible that an abnormal PKD2-driven accelerated degradation of IFNAR1 could neutralize the defensive functions of IFN1 against malignancies. Such scenarios (actual and hypothetical) may include the elevated expression of PKD2, the mutational activation of PKD2, the mutational activation of its upstream regulators, and/or the exposure of cells to PKD2 activators.
PKD2 expression and activities
PKD2 is highly expressed in human prostate cancers and is linked to the invasive characteristics of cells derived from these cancers (Zou and others 2012). Elevated levels of PKD2 correlate with a generally poor prognosis in gastrointestinal cancers (Azoitei and others 2010; Shabelnik and others 2011) and in glioblastomas (Azoitei and others 2011). Intriguingly, a single nucleotide polymorphism within the intron 3 of the Prkd2 gene was reported to be strongly associated with susceptibility to chronic lymphocytic leukemia (Di Bernardo and others 2008). The significance of these findings for PKD2-mediated degradation of IFNAR1 and attenuation of IFN1 signaling in cancer remains to be determined.
Hyperactivation of JAK
Given the role of JAK in IFN1-induced activation of PKD2 (Zheng and others 2011a) and subsequent downregulation of IFNAR1 (Marijanovic and others 2006; Liu and others 2008), it is important to investigate the consequences of elevated JAK activities for IFNAR1 turnover and cellular responses to IFN1. Constitutively active JAK1 and JAK2 mutants have been associated with the development of diverse hematologic malignancies such as polycythemia vera, CML, various types of acute lymphoblastic leukemia, and lymphomas [reviewed in Constantinescu and others (2008); Vainchenker and others (2008); Costa-Pereira and others (2011)]. Even in the absence of JAK mutations, the IFN1-independent induction of JAK by other polypeptide cytokines could hypothetically contribute to the accelerated degradation of IFNAR1 without providing the benefit of any antitumorigenic signaling. These suspect stimuli include hormones [such as growth hormone and prolactin implicated in the pathogenesis of breast cancers (Frank 2008; Swaminathan and others 2008; Wagner and Rui 2008)] and interleukin (IL)-6, which is a key mediator of critical contribution of inflammation to the development and progression of various cancers [reviewed in Grivennikov and others (2010)].
Oncogenes constitutively activating PKD2
PKD2 can be activated by some oncogenic proteins, including BCR-ABL (Mihailovic and orthers 2004) and Src (Storz and others 2003; Storz and Toker 2003; Waldron and Rozengurt, 2000). While the role of Src in regulating IFNAR1 stability and IFN1 signaling remains untested, recent evidence strongly implicates constitutive PKD2 activation by BCR-ABL in the dramatic downregulation of IFNAR1 and concurrent decreased sensitivity of CML cells to IFN1. Importantly, the exposure of CML cells to low doses of imatinib restored both IFNAR1 stability and the sensitivity of CML cells to the antiproliferative and antisurvival effects of IFN1 (Bhattacharya and others 2011b).
Other activators of PKD2
Another important stimulus that activates PKD2, leading to IFNAR1 phosphorylation, ubiquitination, and downregulation, is VEGF, an angiogenic cytokine that is produced abundantly in tumors and plays a key role in tumor development and progression (Ferrara 2004; Selleck 2006). Prior exposure of cells to VEGF leads to the loss of IFNAR1 and the attenuation of IFN1-induced STAT activation (Zheng and others 2011b). Furthermore, cells from mice harboring one allele of the mutant IFNAR1, in which Ser526 within the phospho-degron is substituted with alanine (Ifnar1tm1.1Syfu ), were resistant to VEGF-stimulated IFNAR1 downregulation. Intriguingly, these mice produced a noticeably lesser amount of de novo formed blood vessels in a VEGF-impregnated Matrigel plug. This suggests that the efficient degradation of IFNAR1 and the ensuing restriction of antiangiogenic IFN1 signaling might be important for angiogenesis (Zheng and others 2011b).
The activation of PKD2 was also reported in response to the expression of the NS3 protein of the hepatitis C virus that contributes to the development of hepatocellular carcinoma (Lu and others 2008). Other potentially important mechanisms that activate PKD2 include inducers of the G-protein-coupled receptors [GPCR (Sturany and others 2002)] and the cross-linking of B- and T-cell receptors (Sidorenko and others 1996). The immunosuppressive and protumorigenic role of GPCR inducers such as adenosine (Sitkovsky and others 2008), as well as the important role of lymphocyte receptors in the development of lymphomas, has been documented (Falini and Mason 2002). Future studies may determine how the activation of these pathways will affect the sensitivity of cells to IFN1.
Basal IFNAR1 Elimination and Its Acceleration in Tumors
Early in vitro studies demonstrated that even in the absence of an exogenous ligand, high levels of IFNAR1 are toxic for cells, and that the forced expression of IFNAR1 elicits antiproliferative responses (Colamonici and others 1994). These studies suggested that cells should maintain potent means for keeping IFNAR1 levels relatively low to enable cell survival and proliferation. Accordingly, a robust basal mechanism for IFNAR1 downregulation was later identified (Liu and others 2008). These mechanisms occurred in unstimulated cells independently of the addition of ligand or JAK activities. The basal downregulation of IFNAR1 was linked to a constitutive high level of activity of a protein kinase found in cell lysate. This kinase was capable of phosphorylating the key serine residues within the degron of IFNAR1 (Liu and others 2008). Subsequent biochemical purification and pharmacologic and genetic studies identified CK1α as a basal IFNAR1 degron kinase (Liu and others 2009a).
Despite CK1α being constitutively active, CK1α-dependent phosphorylation of its substrates is often dramatically increased by priming the phosphorylation of a proximal Ser/Thr residue at the −3 position from CK1-targeted phosphor-acceptor [reviewed in Knippschild and others (2005)]. An analysis of the primary sequences of IFNAR1 showed that a highly conserved serine residue (Ser532 in human; Ser523 in mice) is located at this position and may act as a priming phosphorylation site. A set of studies subsequently identified this candidate serine as an important priming site that undergoes phosphorylation in the absence of IFN1 and in a manner that does not require JAK activities. This priming phosphorylation plays an important role in accelerating the basal CK1α-dependent phosphorylation of IFNAR1 degron and in the ensuing ubiquitination and degradation of IFNAR1. Additional studies demonstrated the importance of this regulation in limiting cell sensitivity to future encounters with IFN1 (Bhattacharya and others 2010).
While the nature of a direct priming kinase could still be debated, the essential role of p38α stress-activated protein kinase in priming the phosphorylation of IFNAR1 in response to the UPR inducers has been firmly established (Bhattacharya and others 2011a). Signaling pathways converging on the priming phosphorylation and ligand-/JAK-independent elimination of IFNAR1 were delineated after following 3 types of stimuli, including inflammation, danger- or pathogen-associated molecular patterns, and intracellular integrated stress response.
Damage-activated molecular patterns
The effects of viral infection on priming phosphorylation and ligand-independent degradation of IFNAR1 were initially attributed solely to the induction of UPR and the activation of the PERK kinase (Liu and others 2009b; Diehl and others 2011). Subsequent studies using inactivated viruses, incapable of protein synthesis, eventually led to the identification of danger/pathogen-recognition receptors and their downstream signaling not only as potent inducers of IFN1 gene transcription but also as robust stimulators of priming phosphorylation and the ensuing downregulation of IFNAR1 (Qian and others 2011). Importantly, the tumor microenvironment is loaded with the diverse products of necrotic cell death and various vesicles carrying the activators of these danger-associated molecular patterns (Srikrishna and Freeze 2009) that may temporarily suspend the ability of these tissues to respond to IFN1. The specific role of different activators in controlling the sensitivity of tumors to IFN1 under different stages of tumor development remains to be delineated.
Inflammatory cytokines
The second line of investigation that led to the identification of p38α as a priming kinase and as an important regulator of IFNAR1 stability originated from an observation that some human malignant melanoma cells can secrete a soluble factor that eliminates a receptor in a JAK-independent manner (HuangFu and others 2010). The characterization of this factor, produced by a given melanoma cell line, led to the identification of IL-1α as its major component and to the conclusion that the activation of IL1 receptor→TAK1→p38α signaling acts as a mediator to the accelerated degradation of IFNAR1 (Huangfu and others 2012). Other melanoma cell lines could secrete IL-1β or TNF-α (or other inflammatory cytokines capable of activating p38α kinase), largely to a similar effect of eliminating IFNAR1 and dampening the responses to IFN1, including antiproliferative and proapoptotic effects of IFN-β against human malignant melanomas (Huangfu and others 2012). Importantly, the secretion of inflammatory cytokines by tumor-associated leukocytes has been proposed as a major pathogenic factor in the development and progression of many types of human cancers (Grivennikov and others 2010). Regardless of the cellular sources or specific types of these cytokines, their ability to robustly activate p38α signaling, to trigger elimination of IFNAR1, and to suppress IFN1 responses in the tumor microenvironment might contribute to the mechanisms by which the inflammation promotes tumorigenesis.
Integrated stress response
The existence of ligand-independent phosphorylation, ubiquitination, and degradation of IFNAR1 was first noted in experiments that involved forced overexpression of IFNAR1, most of which did not even undergo maturation and trafficking to the cell surface (Liu and others 2008). A similar strain on cellular protein-folding machinery that causes UPR could occur in response to robust viral protein synthesis in an infected cell (Diehl and others 2011). Indeed, IFNAR1 was rapidly downregulated in cells subjected to viral and pharmacologic inducers of UPR in a manner that required the activation of PERK kinase and the phosphorylation of the eIF2α translation inhibitor. This led to the activation of the p38α kinase and the ensuing priming phosphorylation of IFNAR1 (Liu and others 2009b; Bhattacharya and others 2010, 2011a).
Remarkably, the activation of diverse stress pathways [termed integrated stress response, reviewed in Wek and others (2006)] converges on eIF2α phosphorylation, resulting in the temporal stalling of global translation. Pathways involved in the integrated stress response include the activation of the aforementioned PERK (in response to hypoxia, glucose deficit, and other triggers of UPR), GCN2 kinase (triggered by the lack of essential amino acids), or PKR kinase (eg, in response to extracellular glycosaminoglycans in the renovated tumor extracellular matrix). These pathways, which lead to the phosphorylation of eIF2α, play a key role in tumor growth and metastases. When a tumor's mass accumulation outmatches its blood supply, the activation of PERK and GCN2 (triggered by the deficit of oxygen and nutrients) leads to the phosphorylation of eIF2α and the activation of downstream stress kinase-dependent signaling pathways (Diehl and others 2011). Additionally, the constitutive activation of another eIF2α kinase, PKR, has been reported in some hematologic malignancies (Blalock and others 2010), melanomas, colorectal cancers (Kim and others 2002), as well as in breast cancers (Savinova and others 1999; Kim and others 2000; Nussbaum and others 2003). It would be therefore expected that inducers of integrated stress responses can trigger the elimination of IFNAR1. Indeed, phosphorylation and downregulation of IFNAR1 were observed in cancer cells that constitutively activate PKR (Bhattacharya and others 2012). Similarly, deficit of oxygen or nutrients triggers the downregulation of IFNAR1 in cancer cells and fibroblasts in a PERK/GCN2-dependent manner. Both scenarios of integrated stress response include a dramatic desensitization of cells to IFN1. Conversely, pharmacologic or genetic inhibition of eIF2α kinases sensitized human melanoma and breast cancer cells to the antiproliferative and anti-invasive effects of exogenous or endogenous IFN1 in a manner that depends on the integrity of IFNAR1 (Bhattacharya and others 2012). These results support the hypothesis that the activation of diverse integrated stress kinases followed by IFNAR1 downregulation enables tumor tissues to evade the antitumorigenic effects of IFN1.
Oncogenes
The activation of diverse oncogenes that can signal toward p38α kinase in cancer cells is expected to accelerate the degradation of IFNAR1 and attenuate IFN responses via the priming phosphorylation-dependent mechanism. A number of such oncogene families, whose downstream signaling directly activates p38 kinases, include Ets (Yordy and Muise-Helmericks 2000), RAS, and RAF (Loesch and Chen 2008; Lopez-Bergami 2011). Accordingly, the constitutive activities of these oncogenes should attenuate the ability of cancer cells that harbor these oncogenes to respond to IFN1. Additional mechanisms of desensitization may hypothetically be associated with oncogene-stimulated increases in the rate of protein synthesis (eg, characteristic for MYC oncogenes) and with the ensuing activation of UPR (our unpublished observations). Furthermore, some oncogenes may activate p38 kinase indirectly through stimulating the secretion of inflammatory cytokines (Sparmann and Bar-Sagi 2005; Grivennikov and others 2010). These oncogenic activities altogether are expected to orchestrate the accelerated downregulation of IFNAR1 and the evasion of IFN1 control of proliferation and survival. The role of these mechanisms in tumorigenesis and tumor progression merits further investigation.
Sources and Targets of Cross-Eliminative Signaling in Tumors
As outlined in the previous sections, there is a great deal of diversity in stimuli that can elicit cross-eliminative signaling leading to attenuated responses to IFN1 in tumor tissues. This assortment of stimuli includes those of both intracellular and extracellular origin (Fig. 4). Alterations of intracellular physiology, in response to a deficit of oxygen and nutrients or in response to intrinsic signaling events downstream of mutated and constitutively activated oncogenes (eg, BCR-ABL), initiate the inside-out mode of IFNAR1 elimination. Under this mode, downregulation of IFNAR1 and desensitization to IFN1 effects occur in the same cell in which the cross-eliminative signal is initiated.
Conversely, extracellular stimuli such as VEGF, inflammatory cytokines, or danger-associated molecular patterns can act in an autocrine manner and may also affect neighboring cells that belong to diverse compartments of tumor tissue. Importantly, the extracellular cross-eliminative stimuli rely on their own specific cognate receptors, and, accordingly, altering the elimination of these receptors may in turn determine the stability of cell surface IFNAR1 and the extent of cellular responses to IFN1 (Fig. 4).
Under an actual scenario of a developing tumor, it is likely that both types of sources of cross-eliminative signaling could be simultaneously utilized during different stages of tumor progression. Early genetic events such as the mutational activation of oncogenes and/or inactivation of tumor suppressors can suppress IFN1 effects in cancer cells via a cell-autonomous inside-out signaling mechanism, but also can contribute to the secretion of inflammatory mediators that will contribute to IFNAR1 elimination through an outside-in type of action (Fig. 4). An ongoing tumor growth and increasing respiratory/alimentary demands leading to the integrated stress responses may again utilize these intracellular mechanisms. Subsequent recruitment of bone marrow-derived angiogenic and immune cells could further promote IFNAR1 elimination via the additional secretion of VEGF and inflammatory cytokines. Further receptor loss is expected to be compounded by the formation of a necrotic zone and the release of products of necrotic cell death that would act as danger-associated patterns.
What are the targets of eliminative signaling? Given that IFN1 can act via numerous direct and indirect mechanisms, the overall decrease in sensitivity of tumor tissue to these cytokines may encourage tumor development and progression in several of ways (Fig. 4). Obviously, activation of diverse cross-eliminative cascades alleviates the direct antiproliferative, antimigratory, or proapoptotic effects of IFN1 on tumor cells. In addition, the accelerated degradation of IFNAR1 in endothelial cells and their precursors may relieve the suppressive effects of these cytokines on angiogenesis (Folkman and Ingber 1992). The lack of IFNAR1-mediated suppression of growth and proliferation of tumor-associated fibroblasts can alter tumor architecture and optimize the ability of these fibroblasts to provide a niche for tumor-initiating cells (Borovski and others 2011). Finally, downregulation of IFNAR1 in infiltrating immune cells is expected to frustrate the antitumor immunity.
The immune system follows the elimination-equilibrium-escape mode to suppress the viability/growth of cancer cells while also selecting for resistant subclones that subsequently invade and metastasize (Dunn and others 2006). Given the paramount role of IFN1 in the tumor-specific dendritic cell-mediated recruitment and priming of infiltrating T cells and subsequent tumor elimination (Diamond and others 2011; Fuertes and others 2011), it is plausible that downregulation of IFNAR1 in the tumor microenvironment is essential for the switch from the elimination to the equilibrium/escape phases of cancer immunoediting. Future studies will determine the role of IFNAR1 degradation in tumor stroma and the role of the immune compartments of tumors in the processes of tumor development, progression, and metastases.
Eliminative signaling: role in tumorigenesis?
While the aforementioned studies have yet to be carried out, 2 important and pertinent questions could be posed: does IFNAR1 elimination actually occur, and, if yes, does that matter for the tumorigenesis process? Surprisingly, despite many years of studies on the antitumorigenic effects of IFN1, very few studies focused on the levels of expression of IFN1 receptor chains and the alteration of downstream signaling in clinical human cancers.
Downregulation of the IFN1 receptor was demonstrated in patients suffering from hairy-cell leukemia who underwent IFN1 therapy (Billard and others 1986). A loss of IFNAR1 expression and defective Jak-Stat signaling was reported in some human melanoma cell lines that are resistant to IFN-α-mediated growth inhibition (Wong and others 1997; Pansky and others 2000). A substantial (up to 80%) subset of human gastric cancers exhibited a decreased level of either IFNAR1 or IFNAR2 (Chen and others 2009). Constitutively active BRAF and ensuing high levels of β-Trcp2 E3 ubiquitin ligase often coincided with diminished levels of IFNAR1 in a panel of human malignant melanoma cell lines (Kumar and others 2007b). Low IFNAR1 protein levels and their correlation with poor prognosis were reported in a subset of patients with metastatic melanoma (Messina and others 2008). A noticeably stronger immunoreactivity for IFNAR1 was observed in normal skin cells when compared with melanoma cells from the same patients. Furthermore, in clinical samples of malignant melanoma, levels of IFNAR1 inversely correlated with the status of p38α activation (Huangfu and others 2012). It would be expected that a continuing focus on IFNAR1 levels in diverse types of clinical cancers and an improvement in molecular tools in immunohistochemical detection of IFNAR1 will greatly expand upon these important studies.
At least some of attention deficit in examining the role of receptor levels in human cancer could be attributed to doubts as per the role of this receptor in tumorigenesis. In turn, these doubts might stem from a rather underwhelming and unimpressive (at least, from the point of view of a cancer biologist) phenotype observed in IFNAR1-null mice (Muller and others 1994; Hwang and others 1995). These animals do not exhibit a noticeably high rate of spontaneously arising tumors compared to their wild-type control counterparts. Subsequent studies showed that these mice display a modest increase in the growth of transplantable tumors (Picaud and others 2002), and a somewhat greater rate of developing sarcomas upon receiving injections of methylcholanthrene (Swann and others 2007). Recent evidence from these mice nevertheless points to the importance of IFNAR1 expressed in dendritic cells, in cancer immunoediting (Dunn and others 2005; Smyth 2005), and in the immune rejection of methylcholanthrene-induced sarcomas (Diamond and others 2011). A recent report on experiments on a tissue-specific IFNAR1 knockout also suggests the importance of IFNAR1 for natural killer cell maturation, but not in tumor surveillance mediated by these cells (Mizutani and others 2012).
Intriguingly, the very design of these studies invariably relies on the comparison between IFNAR1-null and wild-type animals. If the phenotypes do not differ dramatically enough, a conclusion that IFNAR1 does not play a role in counteracting various aspects of tumorigenesis is usually proffered. However, there are at least 2 alternative interpretations of these negative or unimpressive results.
One interpretation is that, in some experimental models, the antitumorigenic pathways induced by IFN1 are actively altered downstream from IFNAR1 signaling. For example, it has been shown that IFN1 triggers the activation of the tumor suppressor p53-dependent pathways, and that these pathways can contribute to the cell-autonomous, antiproliferative, and proapoptotic effects of IFN1 (Takaoka and others 2003). Accordingly, the development of tumors in animal models that include targeted or spontaneous inactivation of p53 or its downstream targets may not additionally benefit from the lack of IFNAR1.
While the latter possibility is likely to be limited to the cell-autonomous sensitivity to IFN1, the second alternative explanation also encompasses the effects of IFN1 on benign components of tumor tissue, including endothelial cells, tumor-associated fibroblasts, and infiltrating immune cells. This alternative arises from appreciation for potency and diversity of the mechanisms that are triggered in tumor cells and that robustly downregulate wild-type IFNAR1 (presented in this review). If one or a few of these mechanisms ensure an efficient elimination of IFNAR1, it would render the cells harboring the wild-type Ifnar1 genes functionally equivalent to the cells of an Ifnar1 knockout animal. Accordingly, the lack of phenotypical differences between these 2 genotypes will not be sufficient to discount the importance of IFNAR1 in the inhibition of tumorigenesis.
On the contrary, the multitude and potency of cross-eliminative signaling pathways along with the modest effects of IFNAR1 knockout indicate that the loss of cell surface IFNAR1 that occurs as a byproduct of its accelerated degradation might be of paramount significance to the process of tumorigenesis. In other words, among the events leading to tumor development and progression, downregulation of IFNAR1 in malignant and benign components of tumor tissue and microenvironment could be as common (and, hypothetically, as important) as mutational activation, inactivation of oncogenes, and tumor suppressors.
This hypothesis of course needs to be rigorously tested in an animal model where accelerated degradation and downregulation of IFNAR1 in cancer cells and/or in tumor microenvironment are prevented. A viable mouse mutant that harbors a knock-in of ubiquitination-deficient receptor (Ifnar1tm1.1Syfu ) has recently been developed and used to demonstrate the importance of IFNAR1 degradation for efficient VEGF-stimulated angiogenesis in vivo (Zheng and others 2011b). Experiments using these mice to determine the importance of IFNAR1 elimination in tumor development and progression via models of transplantable tumors and chemical/transgenic carcinogenesis are currently in progress.
Medical Significance of Eliminative Signaling
Given the plethora of eliminative influences and the multitude of pathways leading to the cellular desensitization to IFN1, it is a wonder that endogenous or pharmacologic IFN1 can work at all. At this point, having arrived at a fork in the road, we may either abandon the hope for using IFN1 as an anticancer agent or seriously commit to a paradigmatic shift in our approach. On one hand, IFN1 therapy could be limited to patients whose tumors express relevant levels of IFNAR1. On another hand, focused efforts toward the stabilization of IFNAR1 in other groups of patients can be hypothetically entertained as a therapeutic approach. How would our improved understanding of the mechanisms of IFNAR1 elimination translate to the practical undertakings of clinical medicine? There are several potentially important areas to consider.
Development of a means to stabilize IFNAR1
One idea would be to switch from blindly using ever-increasing doses of exogenous IFN1 to discovery, development, and implementation of small molecules designed to inhibit the eliminative pathways to stabilize IFNAR1 and resensitize cells to IFN1. If successful, this approach might yield both economical and clinical benefits. Treatment with IFN1 is very expensive (Lafuma and others 2001; Lafuma and Grob 2003), and this economic burden might be alleviated by combining IFN1 with inhibitors of IFNAR1 degradation that could allow clinicians to achieve efficacy with a lower dose of IFN1. Another scenario is where the small molecules stabilizing IFNAR1 could activate the tumor suppressor abilities of endogenous IFN1 ligands. Besides optimization of the costs of clinical management, lowering the amount of recombinant IFN1 administered to patients is likely to either diminish or entirely prevent the development of neutralizing antibodies that represent a major problem for the treatment of multiple sclerosis patients with IFN-β (Farrell and others 2012; Strayer and Carter 2012).
Given the prominent role of PKD2 and p38α in eliminative signaling, an ideal IFNAR1 stabilizer could possibly combine the inhibitory effects against both kinases. The multikinase inhibitors [eg, Strumberg and Schultheis (2012)] are the subject of increasing interest among oncologists. Although many p38 kinase inhibitors may stimulate cell growth and survival in vivo, the lack of selectivity that plagues their development as drugs (Wrobleski and Doweyko 2005) may pose additional unexpected benefits in this particular case. A proof of principle in the benefit of combining p38 inhibitor with IFN1 has been reported (Huangfu and others 2012). Recent advancements in the design of novel PKD inhibitors (Evans and others 2010; Monovich and others 2010), some of which show promise in preclinical models of pancreatic and prostate cancers, (Harikumar and others 2010; Lavalle and others 2010) also increase optimism.
Optimized combination of IFN1 with already available drugs
Over the past several decades, numerous studies examined the clinical benefits of combining IFN1 with diverse traditional and novel chemotherapeutics—all with variable (if any) success. Our current understanding of the mechanisms regulating IFNAR1 levels may have shed the light on why some of these combinations had a lesser effect than IFN1 alone. It would be expected that drugs that can cause UPR [eg, proteasome inhibitors such as bortezomib (Mujtaba and Dou 2011)] should trigger the accelerated degradation of IFNAR1 and render tumor cells less sensitive to IFN1. A similar outcome of combining IFN1 with
Conversely, a promising strategy may be to combine IFN1 with already available agents that suppress a context-specific predominant mechanism of IFNAR1 elimination. For example, combining low doses of BCR-ABL inhibitors (such as imatinib) that would stabilize IFNAR1 in CML cells with a subsequent treatment of IFN1 demonstrated a robust synergy (Bhattacharya and others 2011b). These results are clinically relevant for efforts to restore the sensitivity of CML cells to IFN-α. This purported mechanism provides the rationale for a strategy using suboptimal doses of imatinib or another tyrosine kinase inhibitor before IFN-α delivery to increase the sensitivity of patients with chronic-phase CML to IFN-α. Although this approach would be limited to a group of patients that do not yet display a resistance to imatinib, it may have certain advantages for this group. First, the use of low doses of imatinib may prevent the development of resistant Bcr-abl mutations. Second, the resistance associated with the life-long use of imatinib might also be prevented if an optimized IFN-α therapy leads to a stable curative effect that enables the discontinuation of its use (Kujawski and Talpaz 2007). These factors increase optimism of the utility of combining BCR-ABL inhibitors with IFN1 for CML treatment (Kiladjian and others 2011; Simonsson and others 2011).
In a similar approach, one could argue for the possibility of combining IFN1 with PTP1B inhibitors or inhibitors of signaling pathways induced by VEGF and/or development of novel agents that could block PERK and other kinases involved in the integrated stress response. However, rising concerns include the feasibility of suppressing diverse pathways, ability to maintain a reasonable balance between targeted stabilization of IFNAR1, and preserving vitally important physiological processes mediated by these kinases.
Targeting the Side Effects of IFN1
Besides increasing the sensitivity of tumors to IFN1, a different approach aimed at protecting normal tissues from the toxic side effects of this cytokine deserves at least some consideration. The side effects of IFN1 are quite severe. They are poorly tolerated by patients and often become the reason behind the discontinuation of IFN1 therapy (Kirkwood and others 2002). As some putative inducers of eliminative signaling (for example, activators of the GPCR that stimulate PKD2) might act in a tissue-specific manner, it might be possible to specifically downregulate IFNAR1 in normal cells to protect them from IFN1-mediated toxicity. While an increased sensitivity to viral infection might pose a problem, the development of a specific IFN1-derived protein antagonist that still elicits antiviral effects, but prevents inflammatory damage (Pan and others 2008), might solve that problem. Future delineation of the mechanisms that govern the extent and duration of the responses of healthy tissues to IFN1 as well as the development of pharmacologic regulators of this sensitivity is merited.
Conclusions
Future of IFN1-based therapy hinges on identification of clinical situations where the sensitivity to IFN1 is either preserved or restored pharmacologically. Despite considerable progress in delineating the mechanisms that regulate the stability of the IFN1 receptor and the sensitivity of normal and cancer cells to these cytokines, numerous questions regarding the crosstalk of these mechanisms and their role in carcinogenesis remain unanswered. The answers to these questions await future studies that are expected to provide unprecedented insight into the role of Type I receptor signaling in counteracting the mechanisms of tumor development and progression. Such studies may potentially point to novel therapeutic approaches.
Footnotes
Acknowledgments
The author apologizes to those colleagues whose work was not cited due to a lack of space. The author thanks Dr. L. Pfeffer for advice and Mr. Y.S. Fuchs for editorial help. Work in the laboratory of S.Y.F. is supported by the NIH/NCI grants CA092900 and CA142425.
Author Disclosure Statement
No competing financial interests exist.