
Abstract
The recognition that genetic factors influence the heterogeneity of individual responses to medications with respect to both toxicity and efficacy is not new. However, only following dramatic advances in functional genomics during the last decade did the development of so-called personalized medicine become a realistic possibility. Although drug development approaches that integrate pharmacogenetic information about both the protein drug and its protein target appear logical, given the complexity of biological systems, the selection of appropriate biomarkers and the study design remain daunting tasks. Here we present potential applications of pharmacogenetics in the development of recombinant coagulation factors. In addition, we highlight the potential utility of a personalized approach to predicting and eventually circumventing immunogenicity using the recombinant Factor VIII in the treatment of hemophilia A as a model system. The immunogenicity of protein therapeutics is of increasing concern during the development and licensure of biologics and clearly calls for a pharmacogenetic approach. This is because, with immunogenicity, the predicament is not that all patients develop inhibitory antibodies but that some individuals, ethnicities, or other subpopulations have a stronger immunogenic reaction than others do.
Introduction
P
Experience with therapeutic proteins has demonstrated that ADAs can develop against nearly all biologicals and that immunogenicity assessments are now an important component of drug development and licensure (Shankar and others 2007). Immunogenicity is a complex phenomenon with numerous risk factors that may be associated with the product, the patient, or both (Rosenberg and Worobec 2004). The risk of developing ADAs cannot be eliminated and regulatory agencies advocate a risk-management approach to immunogenicity (
A broad survey of the causes and consequences of immunogenicity is beyond the scope of this review but have been considered in detail in previous works (Rosenberg 2004, 2005; Rosenberg and Worobec 2004). Risk factors for immunogenicity may be broadly classified as associated with the product or the patient. In recent years, there has been an increased focus and investments in innovation and streamlining of upstream and downstream processing during the manufacture of therapeutic proteins (Walsh 2010). Coupled with the adoption of emerging strategies such as quality by design (QbD) (Rathore and Winkle 2009), one can expect considerable improvements in product quality and consistency and reduced microheterogeneity. While these advancements continue to improve product quality, they cannot control for the diversity of the patient population. Thus, often the predicament is that a protein therapeutic results in ADAs in some individuals or populations and not in others. Here we discuss the potential pharmacogenetic determinants of immunogenicity and how these may be measured, and present clinical examples where a pharmacogenetic approach to predicting and circumventing immunogenicity may have utility.
Pharmacogenetics and Protein Therapeutics
In the current practice of medicine, which is largely based on population responses (Fig. 1), a cohort of patients are subjected to the same diagnosis, and if they test positive receive the same (or similar) prescription. Complex drug development programs and licensure requirements have evolved to ensure that for most individuals a marketed drug will be both safe and effective. However, there will always be a few individuals for whom the drug is unsafe or ineffective or even both unsafe and ineffective (Fig. 1). This is vividly illustrated in an influential study by Spear and others (2001) which showed that the response rates of patients to medications from different therapeutic classes ranged from ∼80% (analgesics) to ∼25% (oncology). Observations that individuals respond differently to an identical medication are not new and date back more than half a century (Kalow 1956). However, only with more recent advances in genomics did the possibility of bringing so-called personalized medicine to a clinical setting become a realistic possibility (Evans and Relling 1999).

Personalized medicine and medical practice based on population responses. A cohort of potential patients is evaluated using the same diagnoses and those that test positive are all given the same prescription (left grey rectangle). The individual responses will, however, vary. For most patients the diagnoses will be accurate and the medication safe and effective (black). However, due to individual genetic variations, the drug will be ineffective for some patients (dark gray), unsafe for some patients (gray), or even both unsafe and ineffective (light gray). Personalized medicine exploits emerging areas of study such as pharmacogenomics to identify the individuals who will either not benefit or suffer adverse consequences with the use of a therapeutic. While the creation of therapeutics unique to every patient may be impractical, therapeutic options can also be designed for subpopulations with unique genetic characteristics.
Supplementary Table S1 (Supplementary Data are available online at
The Pharmacogenetic Basis of Immunogenicity
Immunogenicity assessment represents an important aspect of the development of therapeutic proteins that could benefit from pharmacogenetic considerations (Yanover and others 2011). There has been a transition from the use of proteins purified from animal and human sources to the manufacture of human proteins by recombinant DNA technology. Nonetheless, even when an identical human therapeutic protein is infused into different individuals, some develop inhibitory antibodies and others do not.
Three critical pharmacogenetic parameters that can explain the individual differences in immunogenicity are as follows (Fig. 2):

Immunogenicity risk may be amenable to a pharmacogenetic approach. (1) When an infused therapeutic protein has an endogenous component, the endogenous protein synthesized by the patient is recognized as “self” and provides tolerance. Thus, sequence differences between the endogenous and infused proteins constitute a necessary but not sufficient condition to elicit an immune response. (2) Additional requirements include the generation of the foreign peptides and their presentation by major histocompatibility complex class II (MHC-II) proteins. This adds 2 individual variations to the immune response. First, variations in the proteolytic enzymes or machinery of individual patients determine whether or not the foreign peptides will be generated. Secondly, even if the peptides are generated they will bind with varying affinities to the MHC-II variant of the patient. (3) Peptide-MHC-II affinity and half-life are surrogate markers for immunogenicity. Moreover, MHC proteins are among the most polymorphic in the human genome. This has several consequences. As stated in (2) above different individuals who generate the same foreign peptide may or may not elicit an immune response based on the affinity of their MHC-II variant for that peptide. During drug development, the distribution of different MHC-II variants in the population is also important. For example, a synthetic foreign sequence introduced as a linker or junction during protein engineering may bind with high affinity only to some MHC-II proteins. Not all MHC proteins occur at the same frequency, thus binding of a foreign peptide to a very rare MHC protein will not be as consequential as that of a foreign peptide that binds to a common MHC protein. Finally, MHC proteins do not occur at the same frequencies in all populations. The graphs on the left show the frequencies of MHC-II DRB1* alleles in the North American population (x-axis) plotted against the frequencies of the same alleles in the European, African, and Japanese populations (y-axis). This means that it is plausible that an engineered drug may elicit an immune response in a larger proportion of one population compared to another.
(i) The sequences of the infused protein drug and that of the equivalent endogenous protein (if any) synthesized by the patient. An endogenous source of the infused therapeutic, although a nonfunctional protein, will provide immunological tolerance. Thus, for replacement therapies, the occurrence or lack of endogenous protein in a patient's plasma, that is, the CRIM (cross-reactive immunological material) status of a patient, has been associated with the development of ADAs (Wang and others 2008a). Comparison of the amino acid sequences of the infused and endogenous proteins permits identification of all areas of sequence mismatch.
(ii) The immune response to therapeutic proteins is mediated by major histocompatibility complex class II (MHC-II) proteins. Peptides from regions of the protein with a mismatch must be generated and bind to the MHC-II proteins. Polymorphisms in the network of genes associated with proteolytic processing of therapeutic proteins are likely to contribute to individual variability in this complex and poorly understood phenomenon. Fortunately, the direct identification of therapeutic-protein-derived peptides presented by a patient's MHC-II proteins is now technologically feasible (Bassani-Sternberg and others 2010).
(iii) The MHC-II repertoire of the individual patient is the third critical pharmacogenetic determinant for immunogenicity as MHC proteins are among the most polymorphic in the human genome. Determining the MHC-II restriction is important in understanding the immunogenicity risk associated with a particular T-cell epitope, as the affinity and stability of the peptide-MHC-II complex for any given foreign peptide has been shown to be an important parameter for the strength of the immune response (Lanzavecchia and others 1992).
The 3 pharmacogenetic criteria, namely, sequences of infused and endogenous proteins, antigens presented by MHC-II proteins, and the MHC-II repertoire, can all be measured/evaluated with current technology. More importantly, in vitro and ex vivo assays as well as computational assessments can be used to estimate the stability of the putative peptide-MHC-II complexes. While definitive studies are underway, emerging reports suggest that these criteria may be useful in assessing the potential immunogenicity risk of a drug product to an individual patient.
The Immunogenicity of Factor VIII: Size Matters
The bleeding disorder Hemophilia-A (HA) is treated with infusions of plasma-derived or recombinant (r) Factor VIII (FVIII) (Mannucci and Tuddenham 2001). The development of ADAs that inhibit FVIII function which occurs in approximately 15%–20% of patients overall is currently the most important impediment to the successful management of this chronic genetic disease (Lillicrap 2010). As is the case with immunogenicity in general, the development of inhibitory ADAs against FVIII is a complex process that involves both product- and patient-related factors (Zhang and others 2009).
Genetic data provide some important clues vis-à-vis individuals who may be at increased risk of developing ADAs. Among all HA patients, the prevalence of ADAs is 15%–20% (Boekhorst and others 2008). However, HA is not caused by a single unique mutation in the F8 gene; defective F8 genes can carry over a 1000 different missense mutations, and have mutations in splice sites, nonsense mutations that truncate the protein, and deletions of varying sizes. The prevalence of ADAs in HA patients with the different types of mutations in the F8 gene is shown in Fig. 3. The data that are from a meta-analysis of different studies that together included 5,385 patients (Gouw and others 2012) illustrate a clear pattern with respect to the genetics of the HA causing mutation and prevalence of ADAs. The more extensive is the disruption in the F8 gene the higher the likelihood of developing inhibitors. As a case in point, there would be minimal sequence mismatch between the infused and endogenous FVIII proteins in a patient who carries a single missense mutation and the patient would have central tolerance to the infused therapeutic protein. On the other hand, a HA patient lacking several exons does not synthesize a significant fraction of the FVIII protein and is more likely to react to the infused FVIII drug as a foreign protein.
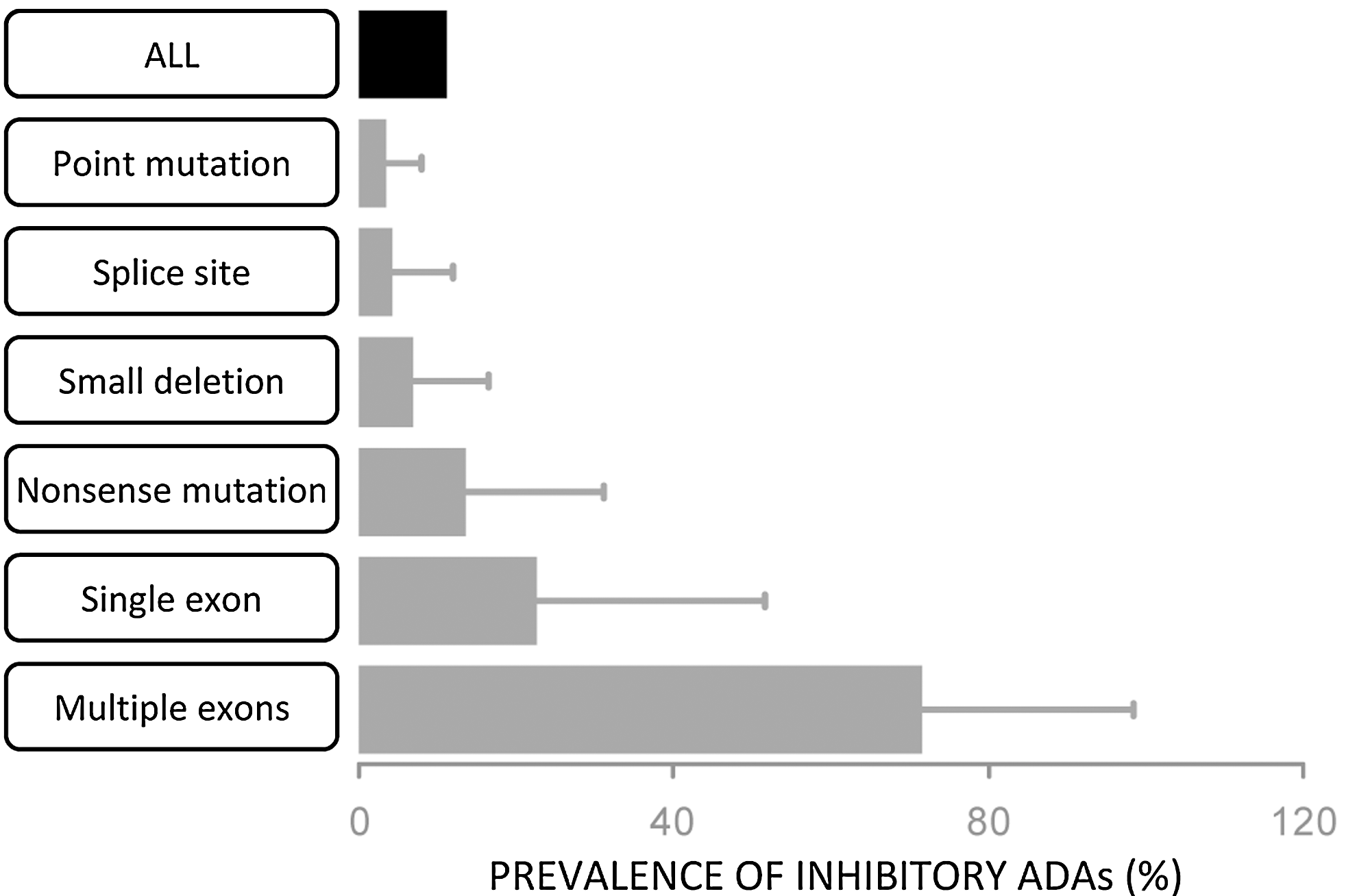
Mutations in the F8 gene of Hemophilia-A (HA) patients and immunogenicity. The graph depicts the prevalence of inhibitory anti-drug antibodies (ADAs) in HA patients with different types of mutations. There is a clear trend that the larger the segment of the endogenous Factor VIII (FVIII) protein that is not expressed the higher the likelihood of developing ADAs. Thus, for example, <5% of patients with a point mutation develop inhibitory ADAs but>70% of patients missing multiple exons do so.
The Immunogenicity of FVIII: Not All Foreign Peptides Are Created Equal
We have seen above that, in general, HA patients with a single amino-acid mutation rarely develop ADAs. This, however, is not true for all missense mutations. Patients with 3 specific mutations at positions 2105, 2150, and 2229 have prevalence rates of ADAs that are 4 to 10 times those observed for all other missense mutations (Astermark and others 2008).
Saint-Remy and co-workers obtained FVIII-specific CD4+ T-cell clones from a patient with one such mutation, R2150H. Characterization of these clones showed that these clones recognized overlapping synthetic peptides with the wild-type sequence, that is, R2150, but did not recognize peptides with the mutant sequence, H2150 (Jacquemin and others 2003). The former constitutes a set of foreign peptides for the patient while the latter are self–peptides, and this suggests that the T-cells can distinguish between self- and wild-type FVIII even when they differ by a single amino acid. The study is also consistent other reports that the anti-FVIII antibodies obtained from HA patients with the R593C and R2150H substitutions recognize the wild-type (foreign) but not the mutant (self) protein (Santagostino and others 1995; Fijnvandraat and others 1997; Gilles and others 1999; Peerlinck and others 1999).
Recent computational advances that combine results of unrelated algorithms have significantly improved estimations of peptides-MHC-II binding affinities (Wang and others 2008b). This consensus method predicts binding in terms of percentile rank, with a low percentile rank reflecting high affinity. We exploited the consensus method to develop an immunogenicty score for each peptide-MHC-II complex (Yanover and others 2011) based on the number of MHC-II protein variants that peptide bound to with very high affinity (percentile score <2). Overlapping peptides that include positions Y2105, R2150, and W2229 exhibit very high immunogenicity scores (Yanover and others 2011). This is consistent with the clinical observation that HA patients with the mutations Y2105C, R2150H, and W2229 have the highest prevalence of ADAs (Astermark and others 2008). Similarly, the percentile scores for the binding of all 15 mer overlapping peptides from the FVIII protein to the MHC-II protein DRB1*1501 (the most common MHC-II allele in the US population) were computed. Only 6 sets of overlapping peptides bind DRB1*1501 with very high affinity (percentile score<2). HA causing missense mutations in 4 of the 6 regions are listed in the Hemophilia A Mutation Database (HADB [
Immunogenicity of FVIII: Underlying Polymorphisms Matter
Recent studies suggest that sequence mismatches between the endogenous (nonfunctional) FVIII of HA patients and the infused protein drug due to nonsynonymous polymorphisms (ns-SNPs) could be risk factors for immunogenicity (Viel and others 2009). There are 4 common ns-SNPs in the FVIII protein of normal individuals and these occur as 5 haplotypes (Viel and others 2007). There are thus 6 variants in the human population, that is, wild-type or one of the 5 haplotypes (Fig. 4). These variants (designated H1 to H6) have gained considerable scrutiny in recent years because a clinical study by Howard and others (2011) led to the hypothesis that these polymorphisms may be associated with higher prevalence of inhibitory ADAs in African American patients (Viel and others 2009). Approximately 90% of Americans of Caucasian descent have the H1 variant and the rest carry the H2 variant, while the variants H1 to H5 were identified in 35.4%, 37.4%, 22.2%, 4%, and 1% of African Americans, respectively. In the clinical study involving 78 African American HA patients, 24% had an H3 or H4 background haplotype and the prevalence of inhibitors was higher among patients with the H3 or H4 haplotypes than among patients with haplotype H1 or H2 (odds ratio, 3.6; 95% confidence interval, 1.1 to 12.3; P=0.04). Importantly, 11 different categories of hemophilic mutation types were identified in the 78 black patients. No difference was observed between the H1/H2 and the H3/H4 haplotype comparison groups in the proportion of patients with higher risk or lower risk types of mutation (P=0.27). These results have given rise to the hypothesis that a mismatch in the sequences of the endogenous (although nonfunctional) FVIII synthesized by the patient and the infused FVIII protein drug could be the underlying cause for the high incidence of inhibitors among black patients (Viel and others 2009; Howard and others 2011; Sauna and others 2012b). This hypothesis has, however, not been experimentally verified and the underlying mechanism for this clinical observation is not understood. The study is also controversial as a similar correlation between sequence mismatch due to polymorphisms and increased prevalence of inhibitors has not been observed in 2 additional studies (Miller and others 2012; Schwarz and others 2012). A large multicenter study involving approximately 1,000 patients of black African descent is currently underway to resolve these issues.
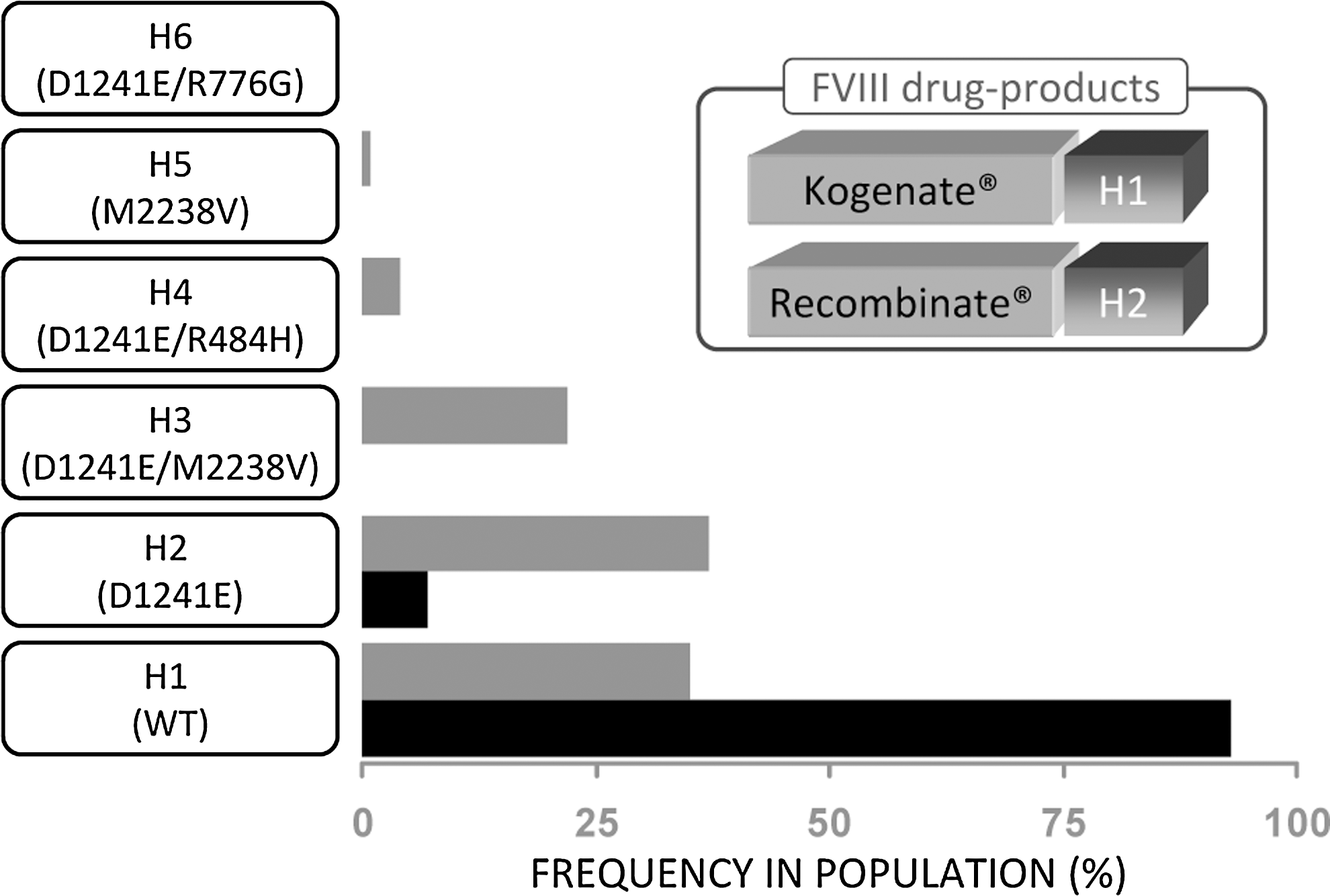
Nonsynonymous haplotypes in the F8 gene. Four common polymorphisms in the F8 gene occur as 5 variants in the human population. The wild-type gene is depicted as H1, while the variants are depicted as H2 to H6. The polymorphisms associated with each variant are depicted on the graph which shows the frequencies of the different variants in the Caucasian (black bars) and African American populations in the US (gray bars). The inset shows that FVIII drug products include both the H1 and H2 variants.
Immunogenicity and Protein Engineering
Most proteins do not make good drug products; thus, the therapeutic protein pipeline now includes a second generation of therapeutics that involves engineering the protein to achieve desirable therapeutic outcomes, patient convenience, or manufacturing efficiencies. Protein engineering provides tools that undoubtedly add considerable value to protein therapeutics; however, the engineered proteins are not free from accompanying risk.
The short- and long-term risks associated with the rapid emergence of fundamentally very different technologies for protein modification can be difficult to predict or even to evaluate. This can be a particular challenge for rare diseases where the clinical trials involve relatively small numbers of individuals. Thus, for example, an engineered Factor VIII protein with the beta-domain-deleted (BDD-FVIII) was approved by the FDA over a decade ago. Nonetheless, the relative immunogenicity of the full-length and BDD-FVIII products still remains controversial. As HA is a rare disease and most studies are relatively small, Alderot and co-workers recently conducted a meta-analysis of prospective clinical trials and suggested that exposure to BDD-rFVIII was associated with an increased risk of all de novo inhibitors (Aledort and others 2011) (for a critique see (Iorio and others 2011).
We have taken a pharmacogenetic approach to this controversy (Sauna and others 2012a). Protein engineering necessarily involves the inclusion of some foreign sequence into a native protein. The BDD-FVIII construct involves the deletion of 894 internal codons and splicing between the third and first nucleotides, respectively, of codons 762 and 1657. Thus, the BDD-rFVIII protein contains a synthetic junction (SFS−QNPPVLKRHQR), formed by the covalent attachment of the 3 N-terminal-most residues of the B-domain, S741F742S743, to the 11 C-terminal-most residues, Q1638N1639P1640P1641V1642L1643K1644R1645H1646Q1647R1648 (Sandberg and others 2001). A computational assessment of the binding affinities of 30 common DR MHC-II isomers for the set of 18 overlapping 15-mer peptides spanning the synthetic BDD-linker showed that the peptides are predicted to bind with high-affinity to only certain DR MHC-II molecules. Approximately 25% of the U.S. population carries the subset HLA-II molecules that bind to the linker peptides. This is a preliminary computational analysis that requires experimental and clinical validation. Nonetheless, it suggests that the immunogenic consequences of bioengineering products should be assessed carefully on an individual patient-by-patient basis. Even if such products do not pose a significantly higher risk than the native products at the population level, they may present a substantial risk to specific individuals. Preclinical evaluation of novel peptides introduced into engineered proteins can also inform clinical studies, for example, ensuring representation of patients with high-risk MHC-II alleles.
The pitfalls of unwanted immunogenicity in bringing bioengineered products to the market are illustrated by the case of vatreptacog alfa. This novel analog of recombinant Factor VIIa carries 3 mutations that resulted in improved thrombin generation and clotting in whole blood from hemophilia patients (Brophy and others 2007). The development of the drug product, however, was halted in September 2012 because some patients developed inhibitors in Phase III trials with the antibodies having a potentially neutralizing effect in one patient [
Future Directions
Immunogenicity is clearly a significant impediment to the optimal use of therapeutic proteins (Shankar and others 2007). The so-called patient related factors have long been implicated as significant risk factors for predicting and/or circumventing immunogenicity. In recent years, there have been noteworthy advances in our understanding of genetic determinants of immunogenicity as well as the development of computational, in vitro and ex vivo methods for the assessment of T-cell-mediated immunogenicity. Simultaneously, there has been a precipitous fall in the cost of sequencing, which now rapidly approaches the 1000-dollar genome mark (Fig. 5). In this review, we have illustrated how genetic markers can aid in assessing the immunogenicity of therapeutic proteins during early stages of drug development. These approaches have the potential to (i) permit the rational development of therapeutic proteins with reduced immunogenicity; (ii) identify individuals, ethnicities, or other populations that are at particular risk of developing ADAs; (iii) design novel therapeutics with reduced immunogenicity for particular at-risk populations; (iv) inform clinical trials by requiring the inclusion of specific genetic markers in addition to race, gender, age, etc.; (v) aid in the design of engineered protein therapeutics with novel junctions, linkers, etc., which potentially have low immunogenicity.
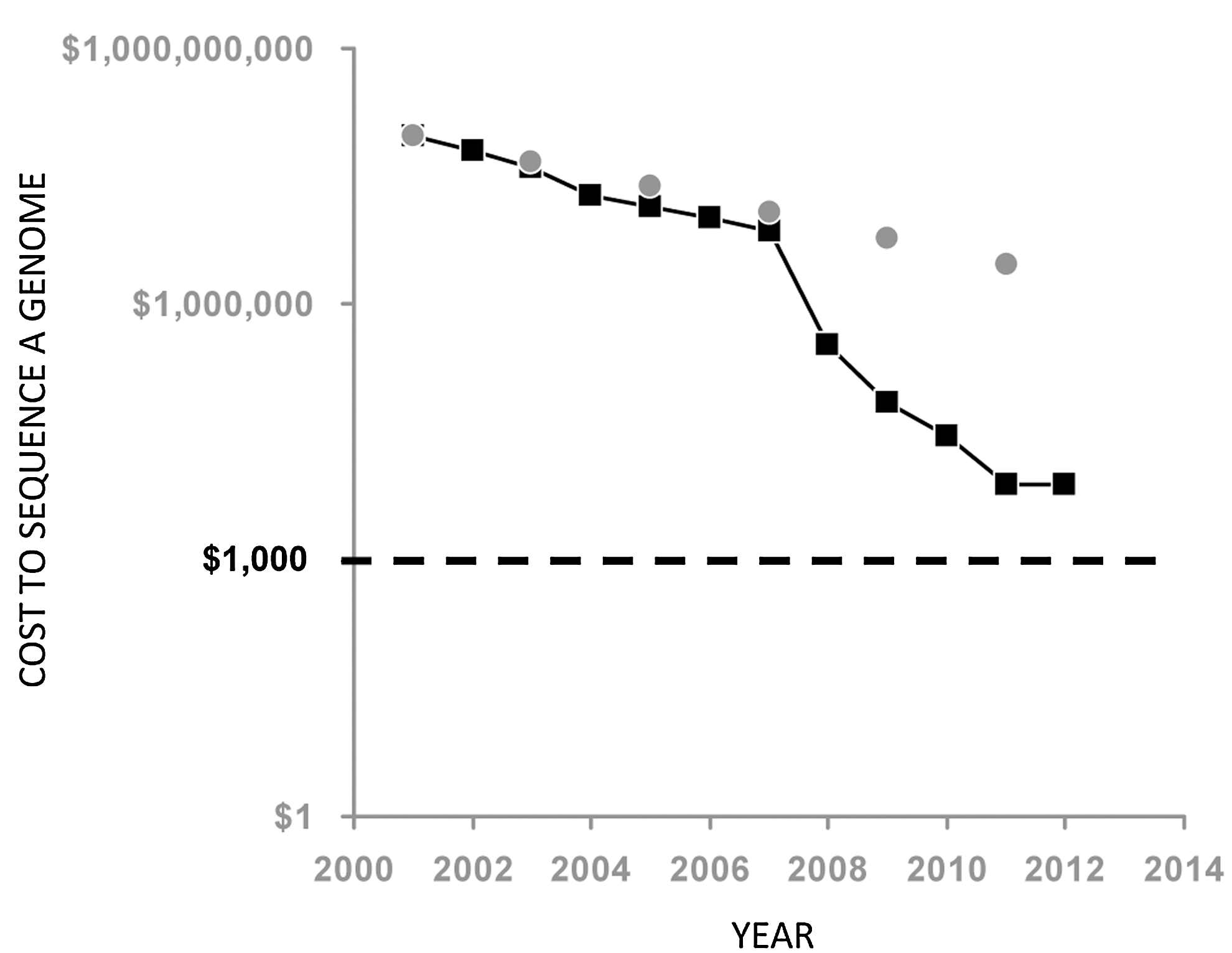
Cost of whole genome sequencing between 2001 and 2012. The cost of whole genome sequencing tracked by the National Human Genome Research Institute (NHGRI) show the precipitous reductions in cost over the last decade (black squares). It is important to note that since about 2008 the reductions in DNA sequencing costs have been considerably greater than what would be expected by Moore's Law (grey circles) which describes a long-term trend in the computer hardware industry that involves the doubling of compute power every 2 years. See
With improvements in manufacturing processes and the adoption of strategies such as QbD (Rathore and Winkle 2009), the quality of therapeutic proteins will continue to improve and product heterogeneity will be reduced. The control of product related risk factors for immunogenicity brings greater importance to pharmacogenetic biomarkers for immunogenicity, which determine why an identical recombinant therapeutic protein is used to treat a cohort of patients some develop ADAs while others do not. The inclusion of a personalized approach in risk-evaluation and risk-mitigation strategies during the development of protein therapeutics, thus, appears to be desirable.
In recent years, there has been a confluence of several technologies that hold promise for the preclinical assessment of immunogenicity risk to individuals or populations with specific genetic characteristics. Considerable effort has been expended to develop computational methods to predict MHC-peptide binding (Wang and others 2008b; Nielsen and others 2010) which allows screening for affinities of large numbers of peptide-MHC complexes at minimal cost. In addition, shortcomings of animal models that limited their utility in immunogenicity assessments are also being addressed. One significant drawback has been the differences in the MHC-II repertoire of humans and animals and thus the use of animal disease models humanized with respect to the MHC-II repertoire holds considerable promise. For example, the hemophilic mouse model that carries the human MHC-II allele, DRB1*1501 (the most common variant in Caucasians), was used to identify peptide regions on the FVIII molecule that contained CD4+ T-cell epitopes (Steinitz and others 2012). These peptide regions could then be screened against a larger repertoire of MHC-II proteins.
Nonetheless, despite unprecedented progress in the emergence of ex vivo, in vitro, and computational methods to assess the immunogenicity of therapeutic proteins, correlations to clinical outcomes are, for the most part, lacking. This remains a major lacuna and requires incentives for the sharing of preclinical and clinical immunogenicity data currently held by individual pharmaceutical companies, patient registries, and academic institutions.
Footnotes
Acknowledgments
We thank Dr. Chava Kimchi-Sarfaty and Dr. Basil Golding for helpful discussions. This work was supported by funds from the Laboratory of Hemostasis and the Center for Biologics Evaluation and Research, Food and Drug Administration's Modernization of Science program (ZES). Our contributions are an informal communication and represent our own best judgment. These comments do not bind or obligate FDA.
Author Disclosure Statement
No competing financial interests exist.