
Abstract
Toll-like receptors (TLRs) are germ-line-encoded innate immune sensors that recognize conserved microbial structures and host alarmins and signal expression of MHC proteins, costimulatory molecules, and inflammatory mediators by macrophages, neutrophils, dendritic cells, and other cell types. These processes activate immediate and early mechanisms of innate host defense, as well as initiate and orchestrate adaptive immune responses. Several single-nucleotide polymorphisms (SNPs) within the TLR genes have been associated with altered susceptibility to infectious, inflammatory, and allergic diseases, and have been found to play a role in tumorigenesis. Critical advances in our understanding of innate immune functions and genome-wide association studies (GWAS) have uncovered complex interactions of genetic polymorphisms within TLRs and environmental factors. However, conclusions obtained in the course of such analyses are restricted by limited power of many studies that is likely to explain controversial findings. Further, linkages to certain ethnic backgrounds, gender, and the presence of multigenic effects further complicate the interpretations of how the TLR SNPs affect immune responses. For many TLRs, the molecular mechanisms by which SNPs impact receptor functions remain unknown. In this review, I have summarized current knowledge about the TLR polymorphisms, their impact on TLR signaling, and associations with various inflammatory, infectious, allergic diseases and cancers, and discussed the directions of future scientific research.
Toll-like Receptors: Sentinel Innate Immune Sensors of Microbial-Associated Molecular Patterns and Endogenous Danger Molecules
TLRs are type I transmembrane glycoproteins expressed by macrophages, neutrophils, DCs, NK cells, and epithelial and endothelial cells (Medzhitov 2007; Kawai and Akira 2011). In addition, T- and B-lymphocytes also express a restricted set of TLRs (e.g., TLR2, TLR7, and TLR9) (Hornung and others 2002; Caramalho and others 2003; Pasare and Medzhitov 2005; Pietschmann and others 2009; Nadigel and others 2011). All TLRs share a common structural organization, expressing an N-terminal ectodomain with multiple LRRs involved in ligand recognition and coreceptor interactions, a transmembrane region, and a C-terminal cytoplasmic tail with the Toll-IL-1R resistance (TIR) signaling domain (Beutler 2009). TLRs sense a diverse array of microbial PAMPs, such as nucleic acids, proteins, lipids, and polysaccharides. In addition, they recognize endogenous danger-associated molecular patterns (DAMPs) [e.g., high-mobility group box protein 1 (HMGB1), host DNA, chromatin], which are normally sequestered in TLR-inaccessible cell compartments, but become exposed to TLRs during inflammation or infection (Kono and Rock 2008; Medzhitov 2009; Seong and Matzinger 2004). TLR2 detects tri- or diacylated lipoproteins from gram-positive bacteria and mycoplasma in association with TLR1 or TLR6, respectively (Lien and others 1999; Ozinsky and others 2000; Bulut and others 2001; Takeuchi and others 2002; Drage and others 2009), as well as senses zymosan (Ozinsky and others 2000), envelope proteins of measles virus (Bieback and others 2002), lymphocytic choriomeningitis, and arena viruses (Zhou and others 2005; Hayes and others 2012). TLR4 is the main sensor for gram-negative bacterial LPS (Medzhitov and others 1997; Poltorak and others 1998; Hoshino and others 1999; Vogel and others 1999), but also detects other structurally unrelated components, for example, mannan (Flo and others 2002; Tada and others 2002), the fusion protein of respiratory syncytial virus (RSV) (Kurt-Jones and others 2000) and chlamydial heat-shock protein (Hsp) 60 (Bulut and others 2002). TLR5 senses extracellular bacterial flagellin (Gewirtz and others 2001); mouse TLR11 protects against uropathogenic Escherichia coli (Zhang and others 2004) and recognizes Toxoplasma-derived profilin-like protein (Yarovinsky and others 2005); and TLR13 senses bacterial RNA and vesicular stomatitis virus (Shi and others 2011; Hidmark and others 2012; Oldenburg and others 2012) (Fig. 1).
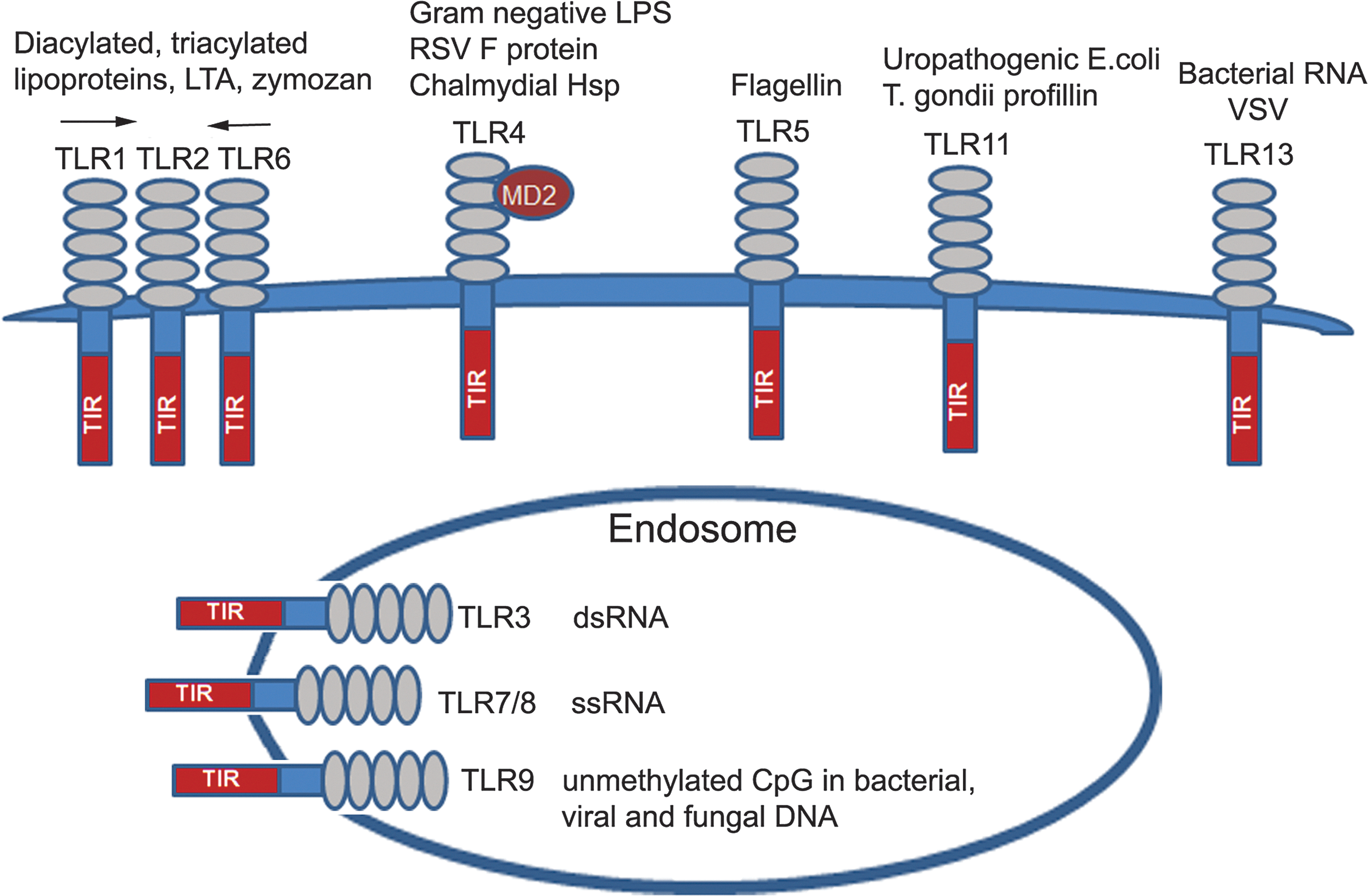
Toll-like receptors (TLRs) and their ligands. TLRs share a common structural organization and consist of an ectodomain with multiple leucine-rich repeats (LRRs) involved in sensing pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), a transmembrane region, and an intracellular tail with the TIR signaling domain. TLRs sense their agonists when expressed on the cell surface (e.g., TLR1, TLR2, TLR4, TLR5, and TLR6), and intracellular TLRs localized to the endosomal compartment detect microbial nucleic acids (see text for the detailed description).
While cell surface-expressed TLRs detect lipids, proteins, and polysaccharides, endosomal TLRs sense microbial nucleic acids; for example, TLR9 recognizes hypomethylated CpG motifs present in the bacterial, viral, and fungal DNA (Hemmi and others 2000; Krug and others 2004; Guggemoos and others 2008; Nakamura and others 2008), whereas TLR3, TLR7, and TLR8 sense viral dsRNA (TLR3) (Alexopoulou and others 2001) and ssRNA (TLR7 or TLR8) (Diebold and others 2004; Heil and others 2004), respectively (Fig. 1). TLRs recognize PAMPs and DAMPs in cooperation with coreceptors, such as CD14 (TLR2, TLR3, and TLR4) (Frey and others 1992; Jiang and others 2005; Lee and others 2006; Akashi-Takamura and Miyake 2008; Zanoni and others 2011) and myeloid differentiation factor 2 (MD2) (TLR4) (Nagai and others 2002). TLR ligation leads to activation of neutrophils, macrophages, and maturation of DCs, upregulates expression of MHC, costimulatory, and accessory molecules, and induces secretion of cytokines and IFNs (Beutler 2009). These processes enhance antigen presentation via MHC molecules and provide the second signal (cytokines and costimulatory molecules) to initiate adaptive immune responses (Medzhitov 2007; Beutler 2009; Schenten and Medzhitov 2011). TLRs expressed in epithelial cells trigger secretion of antimicrobial peptides (Uehara and others 2007; Abreu 2010) and, in endothelial cells, induce expression of the adhesion molecules (Pegu and others 2008).
TLR Signal Transduction
Ectodomains of TLRs recognize PAMPs and DAMPs, either at the concave (TLR4) (Park and others 2009) or convex (TLR2) (Jin and others 2007) interface, in cooperation with the coreceptors CD14, MD2, or CD36 (Tada and others 2002; Jiang and others 2005; Park and others 2009; Stewart and others 2010; Zanoni and others 2011). Agonist recognition initiates TLR homo- (e.g., TLR4) or hetero- (e.g., TLR1–TLR2 or TLR6–TLR2) dimerization that brings together the intracellular TIR domains, forming docking platforms to enable recruitment of adapter proteins and kinases [Fig. 2, rev. in (Vogel and others 2003; Doyle and O'Neill 2006; O'Neill 2008; Beutler 2009]. TLRs use signaling adapters myeloid differentiation primary response protein (MyD) 88 and TIR domain-containing adapter-inducing IFN-β (TRIF) that are coupled to IL-1R-associated kinase (IRAK) 4, IRAK1, and IRAK2 (for MyD88) and TANK-binding kinase (TBK) 1 (for TRIF) (Fitzgerald and others 2003a; Doyle and O'Neill 2006; Li 2008; O'Neill 2008; Carpenter and O'Neill 2009). Upstream kinases become phosphorylated and activated, promoting recruitment and stimulation of downstream adapter-kinase modules that activate mitogen-activated protein kinases (MAPK) and transcriptional factors (Li 2008; Carpenter and O'Neill 2009). Transcription factors translocate to the nucleus and bind to the promoter elements within the target genes, turning on expression of inflammatory mediators, MHC, adhesion, and costimulatory molecules (Vogel and others 2003; O'Neill 2008; Beutler 2009).
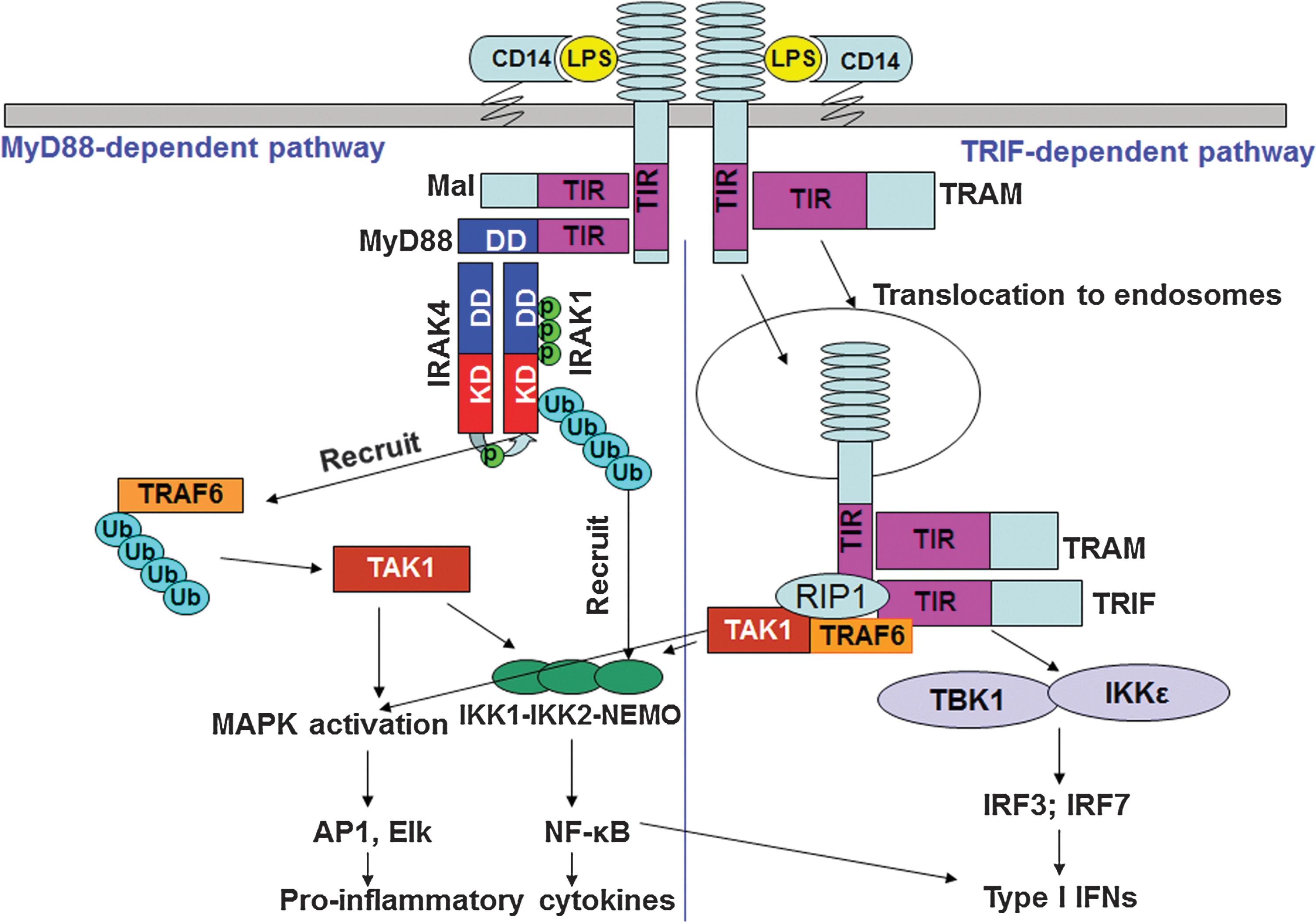
Two TLR signaling pathways. LPS binds to coreceptors CD14 and MD2 that present it to TLR4, causing homodimerization and assembly of TIR domain-containing docking platforms that interact with sorting and signaling adapter proteins. TLR signals via Mal-MyD88 from the cell surface, leading to recruitment and activation of IRAK kinases and engagement of downstream adapters, kinases, and induction of proinflammatory cytokines and chemokines. After translocation to the endosomal compartment, TLR4 associates with TRAM and TRIF, engages TRAF-3, and activates TBK-1 and IKK-ɛ kinases, leading to phosphorylation, activation, and translocation of IRF3. By engaging RIP-1 and TRAF6, this pathway also leads to delayed activation of MAPKs and NF-κB. NF-κB and IRF-3 bind to consensus elements within the interferon (IFN)-β promoter, leading to expression of type I IFN and type I IFN-dependent genes.
TLRs signal via the MyD88-dependent pathway, used by all TLRs except TLR3, and the TRIF-dependent pathway, employed by TLR3 and TLR4 (Vogel and others 2003; O'Neill 2008; Beutler 2009). Figure 2 illustrates utilization of these major signaling pathways by TLR4, as it represents the only TLR using both pathways. TLR2 and TLR4 signal via the MyD88 pathway from the cell surface (Kagan and Medzhitov 2006), although the TLR2/4-MyD88 complexes were also detected in early endosomes and phagosomes (Husebye and others 2010; Mantegazza and others 2012). TLR2/4-associated adapter, MyD88 adapter-like (Mal), acts as a bridging adapter to mediate MyD88 recruitment to the TLR4 receptor signalosome (Kagan and Medzhitov 2006). This forms a scaffold interface that enables recruitment of IRAK4, IRAK1, and IRAK2, whose death domains associate with the MyD88 intermediate and death domains, respectively (Burns and others 2003; Neumann and others 2007). Once MyD88 is recruited to the activated receptor complex, it oligomerizes and recruits IRAK4 to form the MyD88-IRAK4 complex, followed by recruitment of IRAK1 and IRAK2 (Huang and others 2005; Kawagoe and others 2008; Li 2008; Flannery and Bowie 2010; Gay and others 2011). The crystal structure of the MyD88, IRAK4, and IRAK2 death domain complex (Myddosome) revealed a left-handed helix comprised of 6 MyD88 molecules at the bottom, 4 IRAK4 proteins in the middle, and 4 IRAK2 in the top layers (Lin and others 2010). Oligomerization of IRAK4 results in its autophosphorylation and activation of kinase activity, leading to IRAK4→IRAK1/2 phosphorylation and activation of IRAK1/2 (Li and others 2002; Cheng and others 2007). Phosphorylated IRAK1/2 engages a downstream adapter TNFR-associated factor (TRAF) 6 (Dong and others 2006; Keating and others 2007), which is an E3 ubiquitin ligase that undergoes K63-linked polyubiquitination, promoting direct recruitment of transforming growth factor-β-activated kinase (TAK)-1 (Deng and others 2000; Wang and others 2001). IRAK1 also undergoes K63-linked ubiquitination by Pellinos or TRAF6 (Conze and others 2008; Ordureau and others 2008; Moynagh 2009; Xiong and others 2011) that promotes recruitment of inhibitor of nuclear factor-κB (NF-κB) kinase (IKK)-γ via recognition of K63-linked Ub moieties on IRAK1 by the recognition motif within IKK-γ (Conze and others 2008). TAK1 activates MAPKs and the IKK complex, and recruitment of IKK kinases to IRAK1 situates TAK1 and IKK kinases into close proximity, facilitating TAK1-mediated IKK activation (Li and others 2002; Conze and others 2008; O'Neill 2008; Carpenter and O'Neill 2009). IKK-β phosphorylates inhibitor of NF-κB (IκB) proteins, targeting them for K48-linked ubiquitination and proteosomal degradation, leading to the nuclear translocation of NF-κB dimers (Doyle and O'Neill 2006). MAPKs activate transcription factors activator protein-1 and activating transcription factor 2, leading to their nuclear translocation, binding to promoter regions of target genes, and transcription of inflammatory mediators (Vogel and others 2003; Doyle and O'Neill 2006; Medzhitov 2007; O'Neill 2008; Kawai and Akira 2011). TLR7 and TLR9 activate type I IFNs via the MyD88-IRAK4-IRAK1 signaling module, which then interacts with TRAF6, leading to activation of NF-κB and production of proinflammatory cytokines, or activates TRAF3 and IKK-α, leading to induction of IFN regulatory factor (IRF)-3- and IRF7-dependent type I IFNs, respectively (Fig. 2) (Honda and others 2004; Uematsu and others 2005).
Endosome-associated TLR3 signals downstream activation of the NF-κB-dependent and IRF3-dependent cytokine genes via exclusive utilization of TRIF (Kawai and Akira 2011). In TLR4 signaling, the TRIF-dependent pathway is induced from endosomes after endosomal translocation of TLR4 and a sorting adapter, TRIF-related adapter molecule (TRAM) (Kagan and others 2008), enabling recruitment of a downstream adapter TRAF3 (Hoebe 2006; Oganesyan and others 2006). TRIF signals activation of kinases IKK-ɛ and TBK1 that phosphorylate and activate the transcription factor IRF3 and IRF7 (Fitzgerald and others 2003a, 2003b; McWhirter and others 2004; Sato and others 2003; Beutler 2009). IRF3 translocates to the nucleus and, together with NF-κB, activates transcription of type I IFNs (Doyle and O'Neill 2006; Beutler 2009; Kawai and Akira 2011). Apart from induction of type I IFNs, the TRIF-dependent pathway mediates late activation of MAP kinases, NF-κB, and upregulation of costimulatory molecules (Yamamoto and others 2003) (Fig. 2).
TLR trafficking
Trafficking represents an important mechanism responsible for TLR localization to corresponding sites within the cell, which regulates the ability of TLRs to recognize their specific ligands (McGettrick and O'Neill 2010). TLR trafficking is carried out by chaperone proteins gp96, protein associated with TLR4 (PRAT4) A, and Unc93b (McGettrick and O'Neill 2010). Gp96 is an endoplasmic reticulum-associated paralog of the Hsp90 that chaperones several cell surface and intracellular TLRs, as well as other molecules, such as integrins (Yang and others 2007; Wu and others 2012). Deficiency in gp96 leads to the loss of expression of TLR1–5 and TLR7, or improper protein folding (TLR9) (Yang and others 2007), providing multiple TLR knockout phenotypes. PRAT4A is an endoplasmic reticulum-associated protein involved in the trafficking of TLR1, TLR2, and TLR4 to the plasma membrane and mediates trafficking of TLR9 to the endosome/lysosome (Takahashi and others 2007; Liu and others 2010). A multispan transmembrane protein, Unc93b, interacts with intracellular TLRs, such as TLR3, TLR7, and TLR9, and enables trafficking of these proteins to endosomes for signaling (Brinkmann and others 2007). Thus, TLR trafficking is a complex fine-tuning mechanism that controls proper TLR localization, expression, and signaling.
TLR Polymorphisms and Disease
Accumulating evidence supports the role of dysregulated TLR signaling in the pathogenesis of infectious, autoimmune, allergic, inflammatory diseases and cancer (El-Omar and others 2008; Garantziotis and others 2008; Corr and O'Neill 2009; Rakoff-Nahoum and Medzhitov 2009; Netea and others 2012; Theodoropoulos and others 2012). Several synonymous and nonsynonymous single-nucleotide polymorphisms (SNPs) have been identified in the promoter and coding regions of TLR1, TLR2, TLR4, TLR5, TLR7, and TLR9, and their associations with infectious, inflammatory, and allergic diseases are discussed below. The impact of the TLR SNPs on autoimmunity is a large and complex topic that is beyond the scope of the present review, and readers are referred to a series of excellent previous publications (Radstake and others 2004; Liao and others 2010; Lee and others 2012; Netea and others 2012).
TLR1, TLR2, and TLR6 SNPs
Due to recognized roles of the TLR1–TLR2 and TLR2–TLR6 dimers in sensing gram-positive bacteria, Mycoplasma, and mycobacteria (Lien and others 1999; Means and others 1999; Doyle and O'Neill 2006), many studies analyzed the impact of the TLR1, TLR2, and TLR6 SNPs on susceptibility to infections with these pathogens. The nonsynonymous TLR1 SNPs R80T, N248S, and I602S were linked to candidemia in Caucasians, but not in African-Americans, and their presence underlied impaired TLR1-dependent cytokine responses in monocytes of patients with candida infection (Plantinga and others 2012). The TLR1 polymorphism R80T was correlated with Chlamydia trachomatis infection among women with pelvic inflammatory disease (Taylor and others 2012). The TLR1 gene polymorphisms −7202G, N248S, and the I602S showed associations with circulatory dysfunction among patients with severe sepsis, and correlated with whole-blood hyperinflammatory responses to PAMPs, sepsis-associated multiorgan dysfunction, and acute lung injury (Wurfel and others 2008; Pino-Yanes and others 2010). The TLR1 N248S polymorphism that diminishes TLR1-TLR2 signaling was more frequent (the homozygous phenotype SS), and the heterozygous SN genotype was less frequent in patients with leprosy than in the control subjects (Schuring and others 2009). Differences in the allelic frequency in patients who experienced reversal reactions and/or erythema nodosum leprosum reactions indicate that a TLR1 N248S-linked trait may affect the progression from infection to disease, the disease course, and the risk of debilitating episodes (Schuring and others 2009). The I602S TLR1 SNP has been associated with a decreased incidence of leprosy (Johnson and others 2007), suggesting that Mycobacterium leprae subverts the TLR system as a mechanism of immune evasion. The vaginal carriage of Atopobium vaginae has been linked to the presence of the TLR1 polymorphisms within exon 4 (743A>G) and promoter −7202A>G (Verstraelen and others 2009), whereas carries of the R80>T and N248>S TLR1 polymorphisms and the S249>P polymorphism showed an increased susceptibility to invasive aspergillosis after allogeneic stem cell transplantation (Kesh and others 2005).
Polymorphism TLR2_G2258A, a variant causing decreased lipopeptide-induced signaling, was correlated with an increased risk of asymptomatic bacteriuria in women (Hawn and others 2009b). These data suggest that the TLR2 SNPs may contribute to early in vivo human bladder immune responses before the development of symptomatic urinary tract infections (UTIs). The TLR2 polymorphisms, including an insertion/deletion (Ins/Del) polymorphism in exon 1 and a synonymous polymorphism in exon 3 (rs3804100), have been associated with protection against aggressive periodontitis in a Japanese population (Takahashi and others 2011). In contrast, another study failed to find any statistically significant associations between the carriage of the TLR2 SNPs and generalized aggressive periodontitis (Emingil and others 2007). The reason for such contradictory data is unclear, but could be relevant to a small sample size of the latter report. The −196 to −174 Del/Ins TLR2 polymorphism was reported to affect viral loads and susceptibility to hepatocellular carcinoma in patients with chronic hepatitis C (Nischalke and others 2012). The TLR2 promoter, 16934 (rs4696480), and GT microsatellite intron 2 polymorphisms were linked to an increased risk for spontaneous bacterial peritonitis in liver cirrhosis (Nischalke and others 2011).
The TLR2 polymorphisms R688W and R753Q have been studied most extensively, producing both significant associations with various infectious diseases and the lack of thereof. The R753Q polymorphism has been demonstrated to represent a risk factor for cytomegalovirus disease after liver transplantation (Kijpittayarit and others 2007; Kang and others 2012), nasopharyngeal bacterial colonization in infants (Vuononvirta and others 2011), and higher rates of infection recurrence and initial septic shock in liver-transplant recipients developing gram-positive infections (Lee and others 2011). It was linked to a high risk of development of severe infections in critically ill patients from a surgical intensive care unit (Ahmad-Nejad and others 2011). However, in Korean patients, the presence of the R688W or R753Q TLR2 polymorphisms was not linked to liver cirrhosis in hepatitis B virus (Kim do and others 2010). Both the R677W and R753Q TLR2 polymorphisms were found to occur with higher frequencies in patients suffering from tuberculosis from many ethnic backgrounds, and have been linked to impaired signaling capacities (Kang and Chae 2001; Kang and others 2002; Bochud and others 2003; Ben-Ali and others 2004; Ogus and others 2004; Xiong and others 2012). Studies in Turkish children indicate that the R753Q TLR2 polymorphism influences the speed of progression from infection to tuberculosis (Dalgic and others 2011). Other TLR2 SNPs, for example, the P631H mutation and microsatellite polymorphisms in intron 2, were significantly over-represented in patients with tuberculosis, suggesting an increased risk to disease (Yim and others 2006; Etokebe and others 2010; Motsinger-Reif and others 2010; Xue and others 2010a). In contrast, studies in the Chinese and Indian populations showed no associations between the TLR2 SNPs, risk, and prevalence of tuberculosis (Biswas and others 2009; Xue and others 2010b).
TLR3 SNPs
Different polymorphisms in the TLR3 gene have been associated with either protection or predisposition to certain infectious diseases, but the molecular basis for such different outcomes remains unclear. The 299698T/G, 293248A/A, and 299698T/T SNPs in the exon of TLR3 showed association with Stevens-Johnson syndrome (SJS), mucocutaneous disease in Japanese patients (Ueta and others 2007), and the combination HLA-A*0206 and TLR3 rs3775296T/T showed the strongest correlation (Ueta and others 2012). One TLR3 SNP, rs1879026 (G/T), was found to be significantly linked to increased prevalence of infection with hepatitis B virus (HBV), suggesting that genetic variations in the TLR3 gene could affect the outcome of HBV infection (Al-Qahtani and others 2012). In contrast, rs13126816 and rs3775291 TLR3 SNPs were associated with a reduced incidence of human herpes simplex virus type 2 (HSV-2) infections, suggesting that their presence may confer resistance (Svensson and others 2012). Likewise, GWAS have linked the carriage of the L412F TLR3 polymorphism to conferring resistance against HIV infection in the Italian population (Sironi and others 2012). This TLR3 polymorphism was found to protect against acute graft rejection in adult patients undergoing liver transplantation for hepatitis C virus (HCV)-related cirrhosis (Citores and others 2011), and occurred at a significantly higher frequency in Japanese patients suffering from subacute sclerosing pan-encephalitis than controls (Ishizaki and others 2008). Polymorphism in the −705A/G allele at the promoter region of the TLR3 gene was found to predispose individuals in the Indian population to infection with HCV, and molecular analyses demonstrate the lack of impact of this SNP on TLR3 expression (Medhi and others 2011). In contrast, GWAS in German patients failed to reveal any association of the rs5743305 (T/A) promoter polymorphism and a nonsynonymous rs3775291 (C/T) SNP located within exon 4, with clinical parameters of chronic hepatitis C (Askar and others 2009). These reports suggest that the TLR3 gene variations alter host susceptibility to different infectious and inflammatory diseases, but molecular mechanisms by which they affect innate immune responses are largely unknown.
TLR4 polymorphisms
SNPs in the coding and promoter regions of human TLR4 have been linked to an increased incidence of certain infectious and inflammatory diseases, such as gram-negative infections (Agnese and others 2002) and sepsis (Lorenz and others 2002a), RSV bronchiolitis (Tal and others 2004; Awomoyi and others 2007; Paulus and others 2007; Tulic and others 2007b), disseminated candidiasis (Van der Graaf and others 2006), and inflammatory bowel disease (Franchimont and others 2004). At the same time, their carriage has been associated with a lowered risk of atherosclerosis (Kiechl and others 2002a) and rheumatoid arthritis (Radstake and others 2004). Swedish patients with mild or severe UTI were found to express 8 mutations in the TLR4 promoter region, G4038A, G3612A, G3002A, G2604A, A2570G, G2081A, A2026G, and T1607C (Ragnarsdottir and others 2010), 3 of which were also present in patients with myocardial infarction (De Staercke and others 2007), systemic responsiveness (Michel and others 2003), and altered innate immune responses in patients with UTI (Ragnarsdottir and others 2010). In contrast, other studies found no linkage of the A2570G, T2431C, A2026G, and T1607C TLR4 polymorphisms with the prevalence of myocardial infarction (De Staercke and others 2007) or LPS responsiveness (Michel and others 2003).
Two SNPs in the coding region of TLR4 have been identified that encode aspartic acid-to-glycine and threonine-to-isoleucine amino acid substitutions at positions 299 and 399 within the ectodomain, respectively, D299G and T399I (Arbour and others 2000; Lorenz and others 2002b). The D299G and T399I TLR4 polymorphisms occur with a frequency of 5%–10% in Caucasians (Vogel and others 2005), but are relatively rare in Asian populations (Vogel and others 2005). The presence of D299G was originally associated with blunted responsiveness to inhaled LPS and decreased LPS-mediated production of cytokines by airway epithelial cells and alveolar macrophages (Arbour and others 2000), and increased incidence of gram-negative bacteremia (Agnese and others 2002) and sepsis (Agnese and others 2002; Lorenz and others 2002b). The D299G TLR4 polymorphism was also linked to Mediterranean spotted fever caused by Rickettsia conorii (Balistreri and others 2005), and blunted responses to Rickettsia akari were seen in human embryonic kidney (HEK) 293 cells expressing the D299G TLR4 variant (Quevedo-Diaz and others 2010). The 2 TLR4 SNPs also cosegregate in patients (Tal and others 2004; Van der Graaf and others 2006), leading to the most severe LPS hyporesponsiveness compared to the responses elicited by the single alleles (Rallabhandi and others 2006), and their carriage has been associated with sepsis (Lorenz and others 2002b; Barber and others 2004, 2006), severe RSV infection (Awomoyi and others 2007; Tal and others 2004), tuberculosis (Ferwerda and others 2007; Najmi and others 2010; Pulido and others 2010), and myocardial infarction (Edfeldt and others 2004). Human subjects carrying the T399I polymorphism either exhibit a milder LPS-hyporesponsive phenotype or do not manifest it at all (Arbour and others 2000; Rallabhandi and others 2006; Figueroa and others 2012). The D299G and T399I TLR4 polymorphisms were also linked to premature birth rates in a Finnish population (Lorenz and others 2002a), an increased risk of RSV infections in full-term infants (Tal and others 2004; Awomoyi and others 2007), Crohn's disease in Australian, but not Italian or Ashkenazi Jewish populations (Hume and others 2008; Shen and others 2010), and ulcerative colitis (Shen and others 2010). Most recently, the carriage of D299G TLR4 has been linked to an increased risk of endometriosis (Latha and others 2011), recurrent tonsillitis and/or tonsillar hypertrophy (Liadaki and others 2011), and increased prevalence of infections in patients with cirrhosis (Guarner-Argente and others 2010). Although the carriage of these TLR4 SNPs has been associated with several infectious diseases in select cohorts of patients, they confer protection against atherogenesis and decrease susceptibility to rheumatoid arthritis (Kiechl and others 2002b).
While these data have been confirmed by several independent groups (Barber and others 2004, 2006; Henckaerts and others 2009; Shalhub and others 2009; Van der Graaf and others 2006), other studies failed to find a correlation between the occurrence of the D299G or T399I SNPs with gram-negative bacteremia, sepsis, or infections (Feterowski and others 2003; Nakada and others 2005; Everett and others 2007; Jessen and others 2007; Ahmad-Nejad and others 2011). For example, conflicting data have been published regarding association of the D299G polymorphism with tuberculosis. The presence of the D299G did not show association with susceptibility and incidence of tuberculosis in Gambian, Southeastern Chinese, and Korean populations (Newport and others 2004; Xue and others 2010b). However, HIV-infected patients from Tanzania, as well as Indian, Sudanese, and Caucasian patients expressing this SNP had an increased risk of developing active tuberculosis (Ferwerda and others 2007; Najmi and others 2010; Pulido and others 2010; Zaki and others 2012). These studies suggest that the association between D299G and tuberculosis may depend on prior or current immune deficiencies and ethnic backgrounds. These discrepancies could be due to limited sample sizes and power, analyses of different ethnic populations, and multigenic effects, requiring the need for extensive studies of a larger number of patients with different ethnic backgrounds and genders to draw definitive conclusions.
SNPs in other TLRs
Polymorphisms in other TLRs have been also linked to modified susceptibility to infectious diseases in certain human populations. A polymorphism in the ligand-binding domain of TLR5, 392STOP, was associated with defective responses to flagellin, increased susceptibility to pneumonia caused by Legionella pneumophila (Hawn and others 2003), and an increased risk of UTIs in Caucasian women (Hawn and others 2009a). Further, this polymorphism has recently been linked to bronchopulmonary dysplasia in preterm infants (Sampath and others 2012), exhibited negative correlation with Crohn's disease, but its carriage had no effect on susceptibility to infection with Salmonella enterica serovar Typhi in the Vietnamese cohort (Dunstan and others 2005). The polymorphisms rs10737416 in TLR5, rs6531656 in TLR6, and rs337629 in TLR10 were associated with the occurrence of acute graft-versus-host disease after hematopoietic stem cell transplantation in the Finish population, and the TLR5 SNPs rs2800230 and rs2800237 were linked to chronic disease (Sivula and others 2012).
Variations in the TLR7 and TLR9 genes have shown certain associations with viral and bacterial infections, respectively, and modified responses of patients to therapeutic interventions. For instance, the c.32A>T variation in TLR7 was over-represented in female patients with chronic HCV infection compared to patients with other chronic liver diseases and healthy controls, and its presence had a predictive value of an unfavorable outcome of IFN-α therapy, suggesting that variations of TLR7 alter the immune response to HCV infection (Schott 2008). Likewise, polymorphisms in TLR7 and TLR8 have been linked to susceptibility to HCV infection in the Taiwanese population (Wang and others 2011). On the other hand, the c.1-120T>G TLR7 SNP was found to protect against advanced inflammation and fibrosis in male patients with chronic HCV infection (Schott 2007).
Analyses of HIV-positive patients from the Swiss HIV cohort study revealed that the 1635A/G and +1174G/A TLR9 SNPs in the linkage disequilibrium were associated with rapid progression of HIV infection, highlighting its potential importance for therapeutic targeting and vaccine development (Bochud and others 2007). Further, the carriage of the TLR7 Q11L polymorphism was linked to increased HIV-1 susceptibility, accelerated progression, higher viral loads, and advanced immune suppression in female patients, and patients' PBMCs expressing mutant TLR7 had lower IFN-α release upon TLR7 activation (Oh and others 2009).
TLR Mutations, Allergy, and Inflammatory Diseases
Pathogenesis of allergic diseases is underlied by deregulated expression of cytokines by macrophages and DCs that drive differentiation and regulate the balance of Th1/Th2/Th17 populations (Minnicozzi and others 2011; Holgate 2012). Because TLRs are primary sensors of PAMPs, DAMPs, and stress signals associated with allergen exposure, genetic variations in the TLR genes may influence the incidence, severity, and outcome of allergic diseases. Many GWAS have tested this hypothesis and confirmed the presence of such an association. For example, patients with atopic dermatitis (AD) expressing the R753Q TLR2 variant had severe eczema associated with decreased production of IL-8 from monocytes stimulated with lipoproteins or heat-killed Staphylococcus aureus compared to the responses of monocytes from WT TLR2-expressing AD patients (Mrabet-Dahbi and others 2008). Interestingly, while no difference was found between patients with mutant and wild-type TLR2 for IL-5, TNF-α, IFN-γ, and IL-2 production (Mrabet-Dahbi and others 2008), subsequent studies from the same group demonstrated increased secretion of IL-6 and IL-12 (Niebuhr and others 2008). Further, a more recent report showed a significantly higher frequency of the R753Q TLR2 mutation and different frequency of the D299G TLR4 SNP in Italian children with AD compared with the controls, and association of the R753Q SNP with more severe clinical manifestations (Salpietro and others 2011). Thus, TLR2 SNPs may be essential in the pathogenesis of AD and involved in the enhanced susceptibility to skin infections with S. aureus.
SNPs in the TLR6 gene, rs5743798 and rs6531666, were observed more often in Dutch children with atopic eczema than in control subjects, suggesting that they could alter the Th1/Th2 balance, causing an increased risk of developing atopic disease (Miedema and others 2012). In children with the CD14/-159CC, CD14/-159TT, and TLR9/2848GA genotypes, increased exposure to housedust mites was associated with reduced incidence of allergic rhinitis. Analyses of how housedust mite exposure affects the incidence of atopy in children revealed significant associations with SNPs in the CD14 and TLR4 genes. Children expressing CD14/-159CC and CD14/-1359GG had a significant positive correlation between allergen concentrations in household and sensitization, whereas the carriage of the TLR4/896 AG allele conferred protective effects (Kurowski and others 2011). Thus, development of sensitization and allergy is likely to be linked to the TLR polymorphisms and is modified by allergen exposure.
Since many studies have demonstrated a connection between asthma and the cytokines IL-4 and IL-13 that play an important role in the regulation of the Th1/Th2 balance (Minnicozzi and others 2011), Adjers and others (2005) investigated the impact of the TLR4 and IL-4 polymorphisms on the prevalence of asthma. They found that a combination of the low LPS-responsive allele G of TLR4 and high IgE production allele T of IL-4 underlies an increased risk of asthma in Finish women (Adjers and others 2005). GWAS in the Turkish population revealed that the D299G and T399I TLR4 polymorphisms are associated with mild forms of asthma in atopic asthmatics (Saçkesen and others 2005). In Swedish children, the D299G TLR4 polymorphism was associated with a 4-fold higher prevalence of asthma, but not to allergic rhinoconjunctivitis, as well as with reduced IL-12 p70 and IL-10 production (Fagerås Böttcher and others 2004). In contrast, studies of the Danish patients with asthma showed no associations between the TLR2 or TLR4 polymorphisms and new-onset asthma (Smit and others 2007), whereas Canadian nonsmoking individuals showed a reduced prevalence of hay fever and atopy in carriers of the D299G TLR4 polymorphism (Senthilselvan and others 2008). Similarly, exposure of nonsmoking, nonatopic healthy subjects to a swine confinement facility with high endotoxin levels revealed reduced airway responses in individuals expressing the D299G and T399I TLR4 polymorphisms compared to age- and sex-matched, wild-type TLR4 carriers (Senthilselvan and others 2009). In the Chinese Han population, polymorphisms in TLR4 gene (the TT genotype of rs1927914 and the GG genotype of rs10983755 and rs1927907) were associated with asthma severity, but does not affect the susceptibility to the disease (Zhang and others 2011). Similar results were reported for the Egyptian population, where allele frequencies of the R753Q TLR2 and D299G TLR4 polymorphisms showed no differences between asthmatic children or controls, while the severity of the disease was significantly influenced by the TLR2 or TLR4 SNPs (Hussein and others 2012). In Tunisian children, the TLR9 (the −1237C allele) and CD14 (the −159 C allele) gene polymorphisms were reported to play a role in predisposition to asthma (Lachheb and others 2008). These reports indicate on the complex interactions between gene variations in TLR4 and susceptibility to allergic diseases that depends on ethnic background and age, and could be further modified by genetic variations in other innate immune response genes, requiring multiparametric, large-scale studies in different human populations.
The TLR2 (rs4696480 A) and TLR4 (rs2770150 TC, rs10759931 GG, rs6478317 GG, and rs1927911 TT) SNPs significantly increased the risk of asthma in children caused by exposure to air pollution particles (Kerkhof and others 2010), indicating that variant alleles of the TLR2 and TLR4 genes influence the susceptibility to the adverse effects of traffic-related air pollution on childhood asthma. GWAS analyses in genotyped subjects from the French epidemiological cohort demonstrated association of the TLR2/+596 C allele with an increased risk for asthma (Smit and others 2009), and analyses of the Norwegian cohort demonstrated that type 1 diabetes and allergic asthma are significantly associated with the TLR2 rs3804100 T allele, suggesting that it could represent a susceptibility locus, whereas neither TLR4 nor CD14 SNPs were associated with these diseases (Bjørnvold and others 2009). Studies of children of European farmers revealed that individuals carrying a T allele in TLR2/–16934 were significantly less likely to have a diagnosis of asthma, current asthma symptoms, atopic sensitization, and current hay fever symptoms compared with children with genotype AA (Eder and others 2004). These results suggest that genetic variations in TLR2 could significantly contribute to the susceptibility to asthma. In GWAS of the Danish cohort, SNPs in TLR7, rs179008, and TLR8, rs2407992, showed a strong association with the development of asthma, rhinitis, and AD (Møller-Larsen and others 2008).
However, studies in other ethnic populations failed to detect linkage between the presence of SNPs and predisposition to allergies. For example, PCR-based genotyping analyses of Italian allergic children with eczema or IgE-mediated food allergy as compared to the healthy controls failed to reveal a significant correlation between carriage of the R753Q TLR2 or D299G TLR4 polymorphisms and prevalence or severity of these allergic diseases (Galli and others 2010). The German cohort of carriers of the D299G and T399I polymorphisms showed a nonsignificant trend toward a lower risk of asthma, and a weaker association for wheeze and endotoxin exposure (Werner and others 2003). No associations of the D299G TLR4 polymorphism with severity of atopy in asthma were found in the UK patients (Yang and others 2004) or in Taiwanese children (Hsieh and others 2009). In the Japanese population, several polymorphisms in the 5′-flanking and coding regions of the TLR2, TLR3, TLR4, and TLR9 genes showed no correlation with the development of atopic asthma and total serum IgE levels (Noguchi and others 2004). Interestingly, analyses of the Dutch population with the data-mining approach multifactor-dimensionality reduction method revealed novel, significant gene–gene interactions of SNPs in IL1RL1 and TLR4 in association with the atopy and asthma phenotypes (e.g., sIgE levels to indoor allergens) (Reijmerink and others 2010). Korean bakery workers expressing the TLR4 variants with polymorphisms (−2027A>G and −1608T>C) were reported to be at a lower risk of developing allergic sensitization to wheat flour- and endotoxin-induced respiratory symptoms (Cho and others 2011). Thus, complex interactions between genetic background, ethnicity, and environmental exposures exist, and the need for complex multiparameter analyses of many innate gene polymorphisms has emerged.
TLR Polymorphisms and Cancer
Since pioneering observations of Rudolph Virchow in the 19th century showing that malignant tumors develop in the regions of chronic inflammation, the link between chronic, smoldering inflammation and cancer has been confirmed by many studies [reviewed in (Balkwill and Mantovani 2001; Mantovani and others 2008; DiDonato and others 2012)]. Because TLRs are key players in initiating and orchestrating inflammatory responses, dysregulated activation of signaling circuits caused by genetic variations within TLRs could have significant consequences on the inflammatory processes and cancerogenesis (Rakoff-Nahoum and Medzhitov 2009). Indeed, expression of the TLR polymorphisms modifies susceptibility to cancer and influences outcomes of therapeutic interventions. Analysis of SNPs in TLR2, TLR3, and TLR4 showed statistically significant differences in distribution of inferred haplotypes between patients with cutaneous melanoma and controls (Gast and others 2011). A 22-bp nucleotide deletion (−196 to −174 del) in the promoter of the TLR2 gene and the D299G TLR4 polymorphisms were found to have a significantly higher frequency in breast cancer patients compared to healthy controls, suggesting that they may confer an increased susceptibility to cancer development (Theodoropoulos and others 2012). The TLR2 rs3804099 C/T and rs3804100 C/T polymorphisms showed a close association with susceptibility to hepatocellular carcinoma, whereas the 2 studied TLR9 polymorphisms, rs352139 and rs352140, had no linkage (Junjie and others 2012). Analyses of 9 allelic variants within TLR2 and TLR5 demonstrated that carriers of TLR2 c. −196 to −174 del (ins/del+del/del) had a significantly decreased risk of gastric cancer, whereas TLR5 rs5744174 C carriers (TC+CC) had an increased risk of gastric cancer and Helicobacter pylori infection (Zeng and others 2011). TLR3 rs11721827 was associated with rectal cancer, and TLR3 rs3775292 and TLR4 rs11536898 were associated with colon cancer (Slattery and others 2012).
The −196 to −174 Del TLR2 and T399I TLR4 polymorphisms have been associated with an increased risk of prostate and cervical cancer in a North Indian population, whereas the TLR3 (c.1377C/T) [rs3775290] and TLR9 (G2848A) mutations showed no correlation (Pandey and others 2009; Mandal and others 2012). Similarly, increased frequencies of the T399I TLR4 polymorphism were noted in patients with nasopharyngeal carcinoma, which was associated with increased expression of IL-1α, TNF-α, and IL-10 expression, suggesting that this polymorphism modifies cytokine and chemokine patterns and plays a role in tumorigenesis (Yang and others 2012). Zheng and others (2004) found a positive association between a sequence variant (11381G/C) in the 3′-untranslated region of the TLR4 gene and the risk of prostate cancer in the Swedish population, and the TLR3 and TLR9 polymorphisms were marginally associated with susceptibility to cervical cancer (Pandey and others 2011). On the other hand, 6 SNPs in the 5′-untranslated region and intron of TLR4 were linked to a decreased risk of hepatocellular carcinoma (Minmin and others 2011), and cancer patients expressing the D299G and various TLR2, TLR4, and TLR5 haplotypes showed increased survival (Gast and others 2011). In contrast, analyses of patients with head and neck small cell carcinoma showed that the carriage of the D299G and T399I polymorphisms had a significantly reduced disease-free and overall survival and poor responses to adjuvant therapy and chemotherapy (Bergmann and others 2011).
Thus, while genetic variation within TLRs allows for an intricate repertoire that enables the host to withstand microbial challenges and confers advantageous outcomes on a population level, carriage of certain genotypes could underlie oncogenesis (El-Omar and others 2008). The TLR polymorphisms perspective for oncogenomic investigations can include rs10008492, rs4833103, rs5743815, rs11466657, rs7696175 (TLR1-TLR6-TLR10 gene cluster); rs3804100, rs4696480, −196- −174 del (Delta22), GT-microsatellite polymorphism (TLR2); 829A/C (TLR3); rs5743836, rs352140 (TLR9). Gaining a better understanding of molecular mechanisms by which SNPs may alter expression, trafficking, signal-transducing functions, and signalosome assembly of TLRs should provide further insights how these polymorphisms modify inflammatory responses and, consequently, susceptibility to cancer.
Molecular Mechanisms by Which Polymorphisms Impact TLR Functions
The molecular mechanisms by which the TLR polymorphisms affect receptor functions are incompletely understood, and controversy exists in the literature regarding the impact of SNPs on TLR expression, localization, trafficking, and signal transduction. The I602S, a common TLR1 SNP, is associated with aberrant trafficking of the receptor to the cell surface and diminished responses of blood monocytes to bacterial agonists. When expressed in the heterologous systems, the TLR1 602S variant, but not the TLR1 602I variant, exhibits the expected deficiencies in trafficking and responsiveness (Johnson and others 2007). Polymorphisms in TLR1 (S150G and V220M) and TLR2 (F670L) were associated with impaired cytokine (IL-4, IL-8, IL-10, IL-12, and IFN-γ) responses of DCs to Mycobacterium avium subsp or Mycobacterium-derived components (Bhide and others 2009).
Very little and controversial information has been reported in the literature regarding the impact of the TLR2 and TLR4 polymorphism on expression levels, signalosome assembly, and signal transduction. One study showed no significant changes in the expression levels of TLR2 in healthy volunteers and patients with familial Mediterranean fever expressing WT and R753Q TLR2 (Soylu and others 2011). In contrast, another group found lower α-CD3 Ab-inducible expression of R753Q versus WT TLR2 in T-cells obtained from healthy volunteers or patients with AD, with distinct modulation of the WT and R753Q TLR2 levels upon stimulation with lipoteichoic acid (Mrabet-Dahbi and others 2008). We recently demonstrated that the R753Q polymorphism does not significantly affect TLR2 expression, but changes the electrostatic potential of the TIR domain, resulting in impaired agonist-inducible tyrosine phosphorylation of TLR2, TLR2-TLR6 dimerization, recruitment of MyD88, and suppressed p38 and NF-κB activation and cytokine expression (Xiong and others 2012). Other loss-of-function TLR2 variants, the P681H and P631H TIR, were also found to be expressed at levels similar to WT TLR2 (Xu and others 2000; Etokebe and others 2010), although the P681 polymorphism was associated with slower rates of internalization from the cell surface to endosomes (Etokebe and others 2010).
Arbour and others (2000) originally reported that compromised responses to LPS of lung epithelial cells in human subjects expressing the D299G TLR4 variant is associated with decreased expression levels of the mutant receptor. Likewise, Prohinar and others (2010) found 2-fold reduced expression of D299G/T399I TLR4 versus WT TLR4 upon coexpression with MD2, and these changes to a certain extent paralleled differences in LPS responsiveness. In contrast to WT TLR4, D299G and T399I variants were incapable of efficiently translocating to the cell surface upon RSV infection, leading to decreased NF-κB activation and production of IFNs, IL-8, and IL-10 (Tulic and others 2007a). In contrast, Rallabhandi and others reported that despite equal total and cell surface expression of WT, D299G, or T399I TLR4s in the HEK293T/CD14/MD2 transfectants, the polymorphic TLR4 species were compromised in their ability to elicit NF-κB activation and cytokine gene expression in response to LPS, F protein from RSV or Chlamydia Hsp60 (Rallabhandi and others 2006; Awomoyi and others 2007). PBMCs from individuals expressing the D299G TLR4 variant showed reduced phosphorylation of IκB-α and secretion of IL-12 p17 upon stimulation with LPS from S. enterica serotype Typhimurium compared to the responses seen in cells expressing WT TLR4, whereas the TLR4 expression levels were similar in the 2 groups (Lundberg and others 2008). Our laboratory also found only minimal changes in total and cell surface expression of the D299G and T399I TLR4 variants and their ability to bind LPS and interact with MD-2 compared to WT receptor species (Figueroa and others 2012). Of note, the D299G polymorphism inhibited LPS-induced association of the mutant receptor with adapters MyD88 and TRIF, resulting in suppressed activation of the transcription factors NF-κB and IRF-3, p38 MAPK phosphorylation, and induction of MyD88- and TRIF-dependent cytokines (Figueroa and others 2012). Davoodi and others (2012) showed that the D299G TLR4 variant expressed in the colorectal cancer cell line HCT116 decreased the levels of p-AKT, p-ERK1, and p-IRAK1 and inhibited IL-8 expression. These data suggest that the D299G TLR4 polymorphic variant expressed in epithelial cells alters signaling and intestinal homeostasis, resulting in chronic intestinal inflammation, and may eventually lead to colorectal cancer (Davoodi and others 2012).
An improved understanding of the molecular mechanisms by which these mutations alter TLR signaling is expected to facilitate the development of new therapeutic strategies for treatment of diseases such as systemic inflammatory response syndrome, acute respiratory distress syndrome, and airway diseases.
Conclusions and Perspectives
Many studies have linked genetic variation in TLRs with susceptibility to infectious and inflammatory diseases, but these interactions are very complex and depend on environmental factors, ethnic backgrounds, gender, and the presence of polymorphisms in other immune genes and MHC. Discrepancies noted between different studies could be due to insufficient sample size, age, sex, ethnic, and racial differences, differences in prevalence of infectious agents in case and control groups, differences in immune responses, or differences in stratification, methods of diagnostics of cancer, chronic inflammatory conditions, and others. Future studies on a larger sample size should shed more light on the significance of the TLR polymorphisms for cancer prevention. Further, comprehensive analyses of molecular mechanisms by which polymorphisms alter TLR functions should provide important mechanistic insights with respect to designing new therapeutic intervention strategies. For example, findings of deficient dimerization of mutant TLR molecules could open the way for a rational design of peptide mimics and small molecules capable of optimizing these processes to improve recruitment of adapters and kinases.
One interesting and crucial line of future research would be to gain a better understanding of how the evolutionary processes and selection pressures from pathogens have shaped the spread of the TLR polymorphisms in human populations. Certain TLR polymorphisms are thought to result from positive selection due to their protective effects from infections caused by certain pathogens, possibly explaining different disease susceptibilities among modern human populations. For instance, the D299G TLR4 variant is prevalent in the African population and has been found to protect against mortality caused by Plasmodium falciparum cerebral malaria (Ferwerda and others 2008). However, because this TLR4 species is linked to the severity of gram-negative sepsis, it could account for the lack of its fixation (100% prevalence) in Africa and may have led to its rare occurrence in Europe and Asia (Ferwerda and others 2008). On the other hand, the positive selection of certain TLR1 alleles in Europeans has been suggested to be due to an attenuated inflammatory response and beneficial effects in sepsis (Barreiro and others 2009). Interestingly, analyses of the evolutionary dynamics of the TLR SNPs in humans have led to a hypothesis that intracellular TLR3, TLR7, TLR8, and TLR9 that sense microbial nucleic acids have been under stronger purifying selection, are less redundant, and express more deleterious mutations compared to TLRs expressed on the cell membrane (Barreiro and others 2009; Netea and others 2012). However, accumulating evidence indicates that TLR4 and TLR1 are also under evolutionary pressure, and the TLR1-TLR6-TLR10 cluster has been the target of recent selection among Europeans (Barreiro and others 2009; Netea and others 2012). Future comprehensive analyses of large human populations are needed to address this hypothesis. Ongoing studies are expected to further clarify the role of genetic variation and disease susceptibility in this important class of innate receptors, and provide important clues for therapeutic targeting of TLRs for therapeutic interventions and treatment of infectious and inflammatory diseases.
Footnotes
Acknowledgments
This work was supported by an NIH grant RO1 AI059524 and University of Maryland, Baltimore–University of Maryland, College Park NIH Seed grant.
Author Disclosure Statement
No competing financial interests exist.