
Abstract
Interferon (IFN)-γ is a cytokine with a variety of functions, including direct antiviral activities and the capacity to polarize T-cells. However, there is limited information available about the function of this cytokine in the avian immune system. To gain a better understanding of the biological relevance of IFN-γ in chicken immunity, gain-of-function (upregulation) and loss-of-function (downregulation) studies need to be conducted. RNA interference (RNAi), a technique employed for downregulating gene expression, is mediated by small interfering RNA (siRNA), which can trigger sequence-specific gene silencing. In this regard, sequence specificity and delivery of siRNA molecules remain critical issues, especially to cells of the immune system. Various direct and indirect approaches have been employed to deliver siRNA, including the use of viral vectors. The objectives of the present study were to determine whether RNAi could effectively downregulate expression of chicken IFN-γ in vitro, and investigate the feasibility of recombinant adeno-associated virus to deliver siRNA in vitro as well. Three 27-mer Dicer substrate RNAs were selected based on the chicken IFN-γ coding sequence and transfected into cells or delivered using a recombinant avian adeno-associated virus (rAAAV) into a chicken fibroblast cell line expressing chIFN-γ. The expression of chIFN-γ transcripts was significantly downregulated when a cocktail containing all three siRNAs was used. Expression of endogenous IFN-γ was also significantly downregulated in primary cells after stimulation with a peptide. Further, significant suppression of IFN-γ transcript was also observed in vitro in cells that were treated with rAAAV, expressing siRNA targeting IFN-γ. Off-target effects in the form of triggering IFN responses by RNAi, including expression of chicken 2′,5′-oligoadenylate synthetase and IFN-α, were also examined. Our results suggest that siRNAs selected were effective at downregulating IFN-γ in vitro both when delivered directly as well as when expressed by an rAAAV-based vector.
Introduction
The functional role of cytokines may be studied through gain- and loss-of-function experiments both in vitro and in vivo. A range of loss-of-function techniques are currently available, of which the RNA interference (RNAi) technique is well established. RNAi is a naturally occurring mechanism in plants and animals that has been used as a molecular tool to examine the function of specific genes and their protein products in various biological pathways (Fire and others 1998). RNAi utilizes small interfering RNAs (siRNAs) that can be transfected into cells or delivered via DNA-based vectors, which upon expression are processed intracellularly to 19–21-nucleotide siRNAs (Grimm 2009). siRNAs mimic the cleavage products of double-stranded RNA (dsRNA) and generally bind to, and activate, the RNase III family nuclease Dicer, which processes them into mature siRNA. The mature siRNAs then interact with a multiprotein RNA-induced silencing complex (RISC) and direct sequence-specific, homology-dependent gene silencing through cleavage and degradation of the mRNA. Depending on the length, RNAi can potentially induce off-target effects through host defense mechanisms (Reynolds and others 2006). Detection of foreign dsRNA initiates the transcription of type I IFNs as well as activation of receptors to detect dsRNA, including dsRNA-responsive kinase (PKR) and a family of 2′,5′-oligoadenylate synthetase (OAS) enzymes (Sledz and others 2003).
Several studies have utilized the siRNA approach to study the role of various cytokines. For instance, Sidahmed and Wilkie (2007) demonstrated the use of antisense oligodinucleotides to inhibit the expression of interleukin-10 and IFN-γ in pig cells. In chickens, the RNAi approach has been used to study genes that play crucial roles in developmental processes in the chick embryo (Pekarik and others 2003; Dai and others 2005; Harpavat and Cepko 2006). In addition, RNAi has been used for in vitro downregulation of the immune system genes in chickens, including the inducible nitric oxide synthase gene (iNOS), Toll-like receptor 3 (TLR3), and nuclear factor kappa B (NF-κB) (Cheeseman and others 2008; Karpala and others 2008; Chiang and others 2009). The RNAi technology has also been employed to inhibit virus replication in chicken cells and tissues (Chen and others 2008; Lambeth and others 2009).
RNAi effects can be mediated through direct administration of short interfering RNA (siRNA) or through expression vectors, including plasmid and viral vectors (Hobel and Aigner 2010). For example, lentivirus- and adenovirus-based vectors have been used for delivery of short hairpin RNA (shRNA)- and microRNA-based constructs in various species (Iba 2000; Grimm and others 2005). Successful delivery of shRNA constructs can be achieved using adeno-associated virus (AAV)-based vectors. AAV belongs to the Parvoviridae family, and Dependovirus genus, reflecting AAV dependency on a helper virus for productive infection to occur. Since its discovery, many AAV serotypes have been isolated from human and nonhuman species; however, all serotypes contain a linear single-stranded DNA (ssDNA) genome of ∼5 kb, 2 open-reading frames (ORFs: rep and cap), and inverted terminal repeats (ITRs) (Stilwell and Samulski 2003). The ITRs, which are the only signals required in cis for encapsulation of DNA, are important elements for initiation of viral replication, integration into the host genome, as well as for recombinant virus generation (McCarty and others 2004). Various serotypes can infect cells from specific or multiple tissue sources, which are determined by the capsid serotype. Of all the known serotypes, serotype 2 (AAV2) has been the most extensively studied to date. The avian AAV (AAAV) was isolated from the Olson strain of quail bronchitis virus, an avian adenovirus (Yates and others 1973). AAAV contains an ssDNA and also requires coinfection with a helper virus for productive infection. Sequence analysis of AAAV has revealed a similar genome size and an organization similar to that of other AAVs (Bossis and Chiorini 2003; Estevez and Villegas 2004). In general, the use of AAV is appealing, because they are nonpathogenic, have the ability to infect dividing or nondividing cells, have specific tissue tropisms, remain episomal, and can be easily engineered as a vector (Grieger and Samulski, 2012).
In the present study, we sought to downregulate chicken IFN-γ in cultured cells using RNAi. We first characterized and validated the efficacy of three siRNA sequences that targeted IFN-γ by transfecting IFN-γ-producing cells with siRNA oligonucleotides, and then modified an AAAV vector to deliver IFN-γ shRNA sequences by viral transduction.
Materials and Methods
Cell culture
Human embryonic kidney cells (HEK 293) were maintained in the Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a 5% CO2–air atmosphere. An immortal chicken fibroblast cell line, DF-1, was maintained in the DMEM (Invitrogen) supplemented with 10% FBS, 1% chicken serum (CS), and 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C and 5% CO2. A chicken macrophage cell line, HD11, was maintained in the RPMI 1640 medium (Invitrogen) supplemented with 10% FBS, 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 41°C and 5% CO2.
Isolation and stimulation of spleen mononuclear cells with a peptide antigen
TROVAC-AIV H5™ (Merial Select, Inc.) is a fowl poxvirus-vectored vaccine that expresses the hemagglutinin (HA) antigen of an H5 avian influenza virus. We have previously shown that a 15-mer peptide (hereafter called B7) is able to induce both HA antigen-specific CD4+ and CD8+ T-cell responses in vaccinated specific pathogen-free chickens (Haghighi and others 2009). Chickens were vaccinated subcutaneously at 14 days of age and received a secondary immunization at 21 days of age, with the recommended dose. At 10 days postsecondary immunization, the spleens were harvested from TROVAC-AIV H5-vaccinated birds and mononuclear cells prepared for in vitro stimulation with B7. Briefly, spleen tissue were rinsed 3 times in 1× Hank's balanced salt solution (HBSS) and then minced with sterile scalpels. The tissue was further disrupted with the flat end of a 10-mL syringe plunger and strained through a 40-μm nylon cell strainer to obtain a single-cell suspension. The suspension was then overlaid onto Histopaque-1077 (Sigma) density gradient, and mononuclear cells at the interface were collected and washed twice in 1× HBSS and then suspended in a complete RPMI medium (containing 10% FBS, 2% CS, 0.146 g
Plasmids and siRNAs
Construction of the recombinant expression vector pcDNA:chIFNγFLAG has been described previously (Haq and others 2011). Briefly, the chicken IFN-γ-coding sequence (CDS) was amplified by PCR using complementary DNA (cDNA) as a template. Primers were designed to amplify the CDS in addition to a 1xFLAG tag. Amplified products were then cloned into pcDNA3.1 (Invitrogen). Orientation of the insert was confirmed using restriction digestion, and sequencing revealed that the insert was identical to the Ensembl transcript IFNG_CHICK, and GenBank NM_205149 reference sequences. Three Dicer-substrate RNAs (DsiRNAs) to target chicken IFN-γ CDS were designed and supplied by Integrated DNA Technologies, suspended to 20 μM concentration, and stored at −80°C. Targeted sequences were (5′-3′) (1) GGCGUGAAGAAGGUGAAAGAUAUCA, (2) GCAAGUAGUCUAAAUCUUGUUC AAC, and (3) CGAUGAACGACUUGAGAAUCCAGCG. As a nontarget control, DsiRNA targeting green fluorescent protein (eGFP) sequence (EGFP-S1 DS) was selected for use. As a positive control, the cloned expression vector pcDNA:chIFN-γFLAG was transfected, and as a negative control, a set of polyethylenimine (PEI)-treated cells (without expression plasmid or siRNAs) was run in parallel with all experiments.
Construction of shRNA expression plasmid vector and recombinant AAAV vectors
An siRNA expression cassette with NdeI-linker ends was made by amplifying the chicken U6 promoter region from the plasmid pRFPRNAiC (ARK-Genomics). The siRNA expression cassette consisted of an U6 promoter followed by a sense sequence (corresponding to the IFN-γ-targeted sequence), a 9-nt loop sequence, an inverted antisense target sequence, and a stretch of Ts as a polymerase III transcriptional termination signal downstream of an U6 promoter. Briefly, 10 ng of pRFPRNAiC and specific primers (Table 1) were used for PCR. The PCR conditions were as follows: 1 cycle of 95°C for 5 min, 35 cycles of 95°C for 15 s, 68°C for 15 s, 72°C for 30 s, followed by a final extension at 72°C for 5 min using Platinum Taq polymerase (Invitrogen). The amplified product was first cloned into the pDrive cloning vector (Qiagen). Ampicillin-resistant colonies were verified by restriction enzyme digestion and sequencing using standard procedures.
siRNA, small interfering RNA; AAAV, avian adeno-associated virus; chIFN-γ, interferon-γ.
Vectors for a helper-free recombinant virus production system were generously provided by Drs. C. Estevez and P. Villegas (University of Georgia, GA). Briefly, the CMV promoter and lacZ ORF from the AAAV backbone vector pEVA3V-LacZ were removed by NdeI digestion and replaced with an NdeI fragment containing the U6-shRNA cassette from pDrive. Cloning was done in Escherichia coli GT116, a deletion strain of E. coli specifically engineered to support propagation of plasmid DNA carrying hairpin structures. Kanamycin-resistant colonies were screened by restriction digestion to confirm insert orientation and sequenced bidirectionally to verify the sequence.
Recombinant AAAV generation
Recombinant AAAV (rAAAV) viral particles expressing an individual IFN-γ siRNA (siRNA-1; Table 1) or eGFP sequence were obtained by cotransfecting HEK293 cells with the rAAAV backbone vector along with two packaging plasmids, which included the Rep/Cap genes and E2A, E4, and VA genes. Cells were seeded in 10-cm tissue culture dishes and transfected when 70%–80% confluent by calcium phosphate precipitation. Cell lysates were prepared 72 h post-transfection. The cell suspensions were freeze–thawed 3 times by transferring samples between a dry ice/ethanol bath and a 37°C water bath. The cell lysate was centrifuged for 30 min at 10,000g at 4°C. Subsequently, the supernatant was removed and transferred into 50 mL Beckman ultraclear tubes. The supernatant was recentrifuged for 6 h at 24,000g at 4°C, followed by dilution of the virus pellet overnight at 4°C with 500 μL HNE buffer [50 mM HEPES (pH 7.4), 0.15 M sodium chloride, and 25 mM ethylenediaminetetraacetic acid] per tube. AAAV was concentrated through a 20% sucrose cushion (2:1 ratio) and centrifuged for 6 h at 24,000 g and 4°C. The supernatant was carefully discarded, and the pellet was dissolved gently in 500 μL HNE buffer. rAAAV stocks were DNase-treated, aliquoted, and stored at −80°C.
Cell transfection and infection
DF-1 cells were cultured in 6-well plates (1×106 cells per well) for 24 h before transfection. Cells were transiently transfected using PEI (Polysciences, Inc.) according to the manufacturer's instructions. Cells were transfected with 2 μg pcDNA:chIFN-γFLAG together with IFN-γ siRNAs or nontarget siRNA controls in a serum-free medium and incubated for 6–8 h at 37°C. Pooled IFN-γ siRNA oligonucleotides were tested at 3 concentrations (10, 1, and 0.1 nM each) or 10 nM for eGFP (nontarget). For viral infection, rAAAV:chIFN-γ shRNA from stock solution was mixed with a serum-free medium to result in appropriate viral genomes per cell and added into a pcDNA:chIFNγFLAG-transfected cell culture medium. For experiments involving rAAAV:chIFN-γ shRNA, viral doses ranged from 3×103 to 30 viral genomes per cell. Subsequently, the transfection medium was removed; cells were washed with phosphate buffered saline; and a fresh culture medium was added to the cells. Cells were collected 24 and 48 h postinfection for RNA extraction. An rAAAV expressing a sequence targeting eGFP was used as the negative control. For the eGFP siRNA group, viral doses of 3×103 viral genomes per cell were used. The presence of rAAAV in cells was detected using primers to amplify a noncoding stuffer region between the ITRs and the coding cassette.
Nucleic acid isolation, reverse transcription, and quantitative real-time PCR
To measure expression levels of various genes, total RNA was harvested from DF-1 cells using TRIzol reagent (Invitrogen) according to the manufacturer's protocol with the addition of 10 μg of glycogen (Invitrogen). Total RNA was then treated with DNase using the DNA-free kit (Ambion). cDNA was prepared from 1 μg of DNase-treated total RNA by reverse transcription using MMLV reverse transcriptase and oligo(dT)12–18 primers (SuperScript™ First-Strand Synthesis System; Invitrogen) according to the manufacturer's instructions. Real-time RT-PCR was performed in a LightCycler 480 instrument (Roche Diagnostics) in a reaction volume of 20 μL using SYBR Green 1 Master Mix (Roche Diagnostics). In addition, the reaction mixture consisted of 0.25 μM of each primer and 5 μL of 1:10 dilution of cDNA. The PCR conditions included a denaturing step at 95°C for 10 min, and subjected to 40 cycles of 95°C for 10 s, 60°C for 5 s, and 72°C for 10 s, followed by 72°C for 5 min. The PCR-positive control consisted of 10 ng of purified pcDNA:chIFN-γFLAG as a template. Expression levels were normalized to β-actin. The ratio of chicken IFN-γ and β-actin mRNA expression was used to calculate relative reduction of IFN-γ expression.
Chicken IFN-γ enzyme-linked immunosorbent assay
The concentration of IFN-γ in cell culture supernatants was assessed using a commercial chicken IFN-γ enzyme-linked immunosorbent assay (ELISA) (Invitrogen) according to the manufacturer's instructions. The concentration of IFN-γ was calculated based upon comparison with a standard curve generated with known amounts of recombinant chicken IFN-γ protein.
Data and statistical analysis
Data were collected and expressed as mean relative expression to normalized β-actin or, for the siRNA experiments, were converted to a percentage relative to the positive control group. Results are depicted as the mean±standard error of the mean of 3 independent experiments. Significance of difference between the means was determined using the Student's t-test in GraphPad Prism software (GraphPad software), with P≤0.05 taken to indicate the significance between treatments.
Results
Silencing of IFN-γ expression using RNAi in vitro
To evaluate the inhibitory effect of RNAi on chicken IFN-γ gene expression in vitro, we cotransfected DF-1 cells with pcDNA:chIFN-γFLAG and a pool of 3 siRNAs that target the IFN-γ CDS. Briefly, the chIFN-γ CDS was FLAG-tagged and cloned into the pcDNA3.1 expression vector and used to express recombinant chIFN-γ (Haq and others 2011). IFN-γ expression in cells transfected with pcDNA:chIFN-γFLAG was compared against cells cotransfected with the pooled IFN-γ siRNA oligonucleotides at 3 concentrations (10, 1, and 0.1 nM each) or 10 nM eGFP (nontarget) control siRNA. Cotransfection with the pool of siRNA at 3 concentrations demonstrated significant reduction in IFN-γ expression using 10 nM at all 3 time points (P≤0.05). However, using 1 and 0.1 nM, a significant reduction was observed only at 24 and 48 h post-transfection (Fig. 1A). We then examined a potential nonspecific host IFN response associated with administration of different concentrations of siRNA (Sledz and others 2003). This was done by examining the expression OAS and IFN-α (data not shown). OAS is a dsRNA-dependent synthetase that activates endoribonuclease RNAse L to degrade ssRNA (Player and Torrence 1998). There was no significant difference in expression of OAS among groups, confirming that the siRNA oligonucleotides did not induce IFN response in a wide range of concentrations (Fig. 1B).
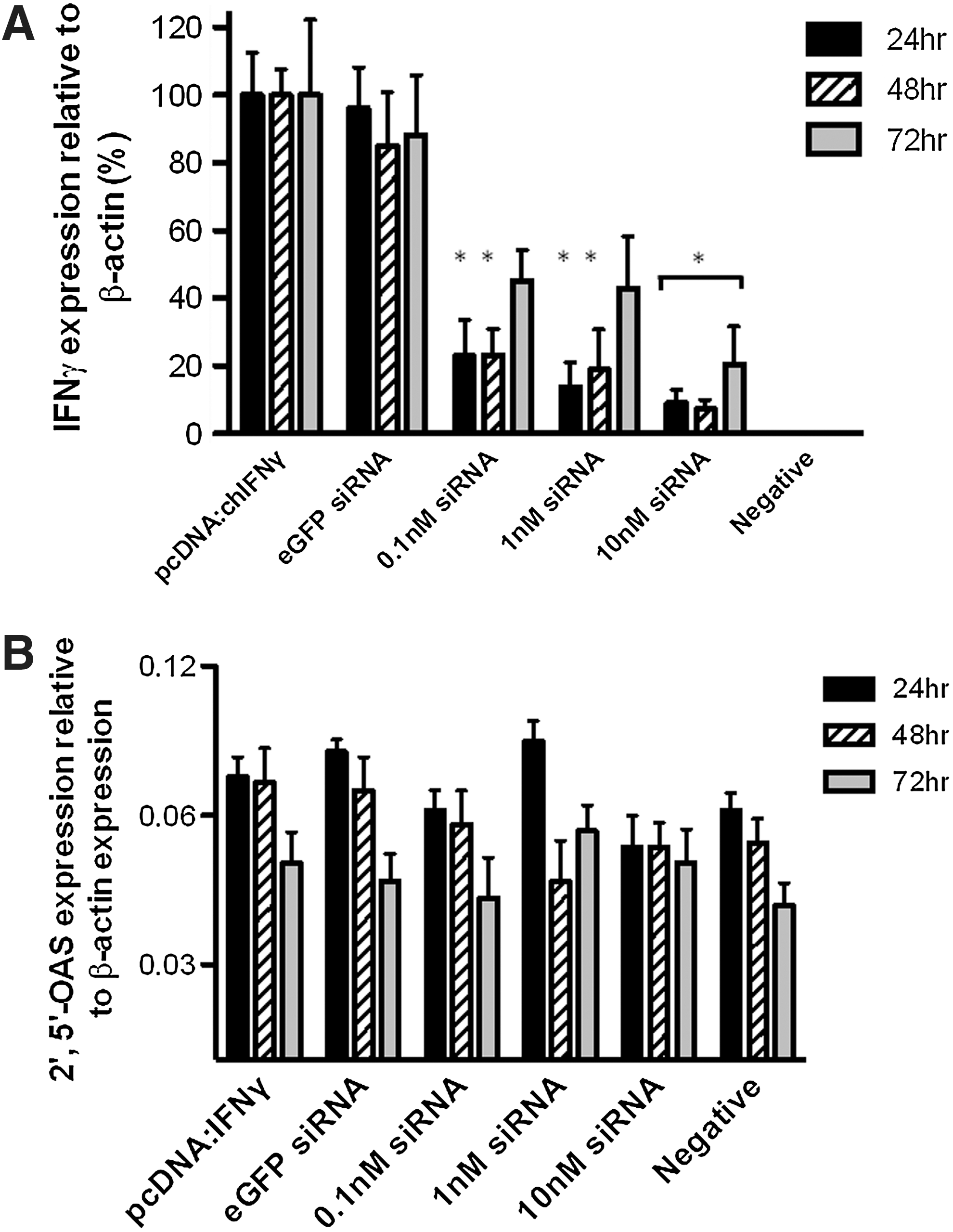
Characterization of in vitro gene silencing by small interfering RNAs (siRNAs) targeting chIFN-γ. DF-1 chicken fibroblast cells were cotransfected with a plasmid encoding chicken interferon-γ (IFN-γ) and siRNAs targeting IFN-γ or nontarget (eGFP) control siRNA. Cells were cultured for varying amounts of time and sampled for RNA expression.
To confirm whether reduced mRNA corresponded to reduced protein, chicken IFN-γ in the culture supernatant was measured by ELISA. Significantly, lower amount of rChIFN-γ was seen at 24 and 48 h post-transfection when compared to both the nontarget and plasmid-only samples. The reduction in the amount of IFN-γ at 72 h in the siRNA group approached significance when compared to the amount of IFN-γ in the nontarget and plasmid-only groups (P=0.051 and 0.052, respectively) (Fig. 2).
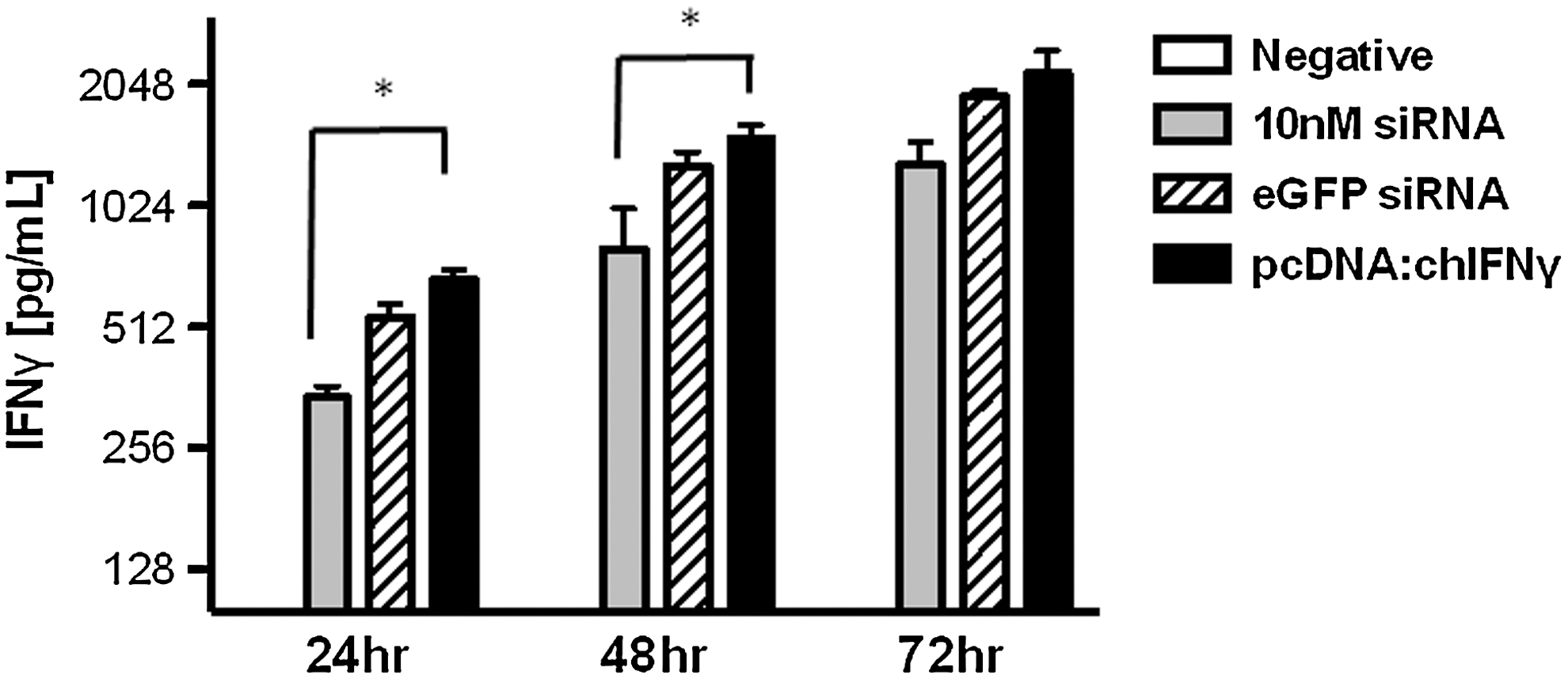
Gene-silencing efficacy of siRNAs targeting chIFN-γ. DF-1 cells were cotransfected with a plasmid expressing chicken IFN-γ and siRNAs targeting IFN-γ or nontarget control siRNAs (eGFP). IFN-γ protein concentrations in the supernatant of transfected cells as determined by enzyme-linked immunosorbent assay. Supernatants were collected 24, 48, and 72 h post-transfection. Experiments were done in triplicate. Error bars represent the mean±SEM, and * denotes significance (p≤0.05) when compared to non-target and positive controls. Negative refers to supernatant from mock transfected cells.
Silencing endogenous IFN-γ expression using primary cells
Spleen cells from chickens vaccinated with TROVAC-AIV H5 are able to induce high levels of IFN-γ expression in response to a 15-mer peptide (B7) from the HA antigen of avian influenza virus (H5-AIV) (Haghighi and others 2009). To investigate the ability to knockdown endogenous IFN-γ mRNA using these siRNAs, spleen cells from vaccinated birds were stimulated in vitro with B7 and treated with siRNAs. Expression levels of IFN-γ confirmed the ability of these siRNA to significantly knockdown expression of IFN-γ at both 12 and 24 h (Fig. 3A). Reduced levels of IFN-γ protein in the supernatant of B7-stimulated primary cell-transfected siRNA was confirmed by treating HD11 cells with the culture supernatants and measuring NO production using the Griess assay (Fig. 3B).

Quantitation of siRNA-induced knockdown of endogenous chIFN-γ in primary spleen cells. Spleen mononuclear cells from 4-week-old chickens, vaccinated with a fowl poxvirus-vectored vaccine expressing the hemagglutinin (HA) antigen of avian influenza virus, were harvested and incubated with a peptide derived from the HA antigen along with control (eGFP) or IFN-γ-specific siRNAs. Expression of endogenous chIFN-γ in primary spleen cells
In vitro evaluation of IFN-γ mRNA downregulation by rAAAV
Based on a pilot study comparing the silencing efficiency of the 3 siRNAs (data not presented), one was selected (siRNA-1) for cloning into an rAAAV. The eGFP siRNA sequence was used as a nontarget control. The chicken U6 promoter was used to drive expression of shRNAs in chicken cells.
To evaluate the ability of rAAAV-encoding chIFN-γ shRNA to downregulate expression of IFN-γ, the virus was added to chicken fibroblast cells (DF-1) previously transfected with a plasmid containing the CDS of chicken IFN-γ. After addition of rAAAV to the culture, cells were monitored for cell death. No cell death was observed among all cells infected with rAAAV. Infection by rAAAV was confirmed by amplifying a noncoding stuffer region located between the viral ITRs from cellular DNA. No viral product was detected from untreated cells (data not presented).
We next measured the levels of IFN-γ transcript abundance by quantitative RT-PCR after rAAAV infection. A reduction of 50%–80% in IFN-γ transcripts was observed at 24 and 48 h postinfection compared to uninfected cells expressing exogenous chIFN-γ. In contrast, levels of IFN-γ mRNA were not significantly changed after infection with rAAAV targeting a nonphysiological target gene (eGFP). Further, we demonstrated a dose–responsive decrease in IFN-γ transcript levels with increasing viral titer (Fig. 4). Thus, rAAAV is a suitable vector for gene silencing in the chicken model.
Recombinant avian adeno-associated virus-mediated knockdown of chIFN-γ. IFN-γ expression relative to the pcDNA:chIFNγ-treated DF-1 cells (%), at 24 and 48 h postinfection. Bars are the average of the triplicate experiments±the SEM. *=Significant when compared to pcDNA:chIFNγ- or rAAAV:eGFPshRNA-infected groups (P≤0.05).
Discussion
The present study demonstrated the successful knockdown of chIFN-γ using siRNA molecules designed to target the CDS of this cytokine. The host recognition of invading pathogens or response to vaccines may trigger the secretion of cytokines that play critical roles required for elicitation and regulation of the host immune response. A better understanding of the role of these cytokines in immunity requires loss-of-function studies. Application of RNAi, which can be induced by siRNA or shRNA molecules, is an approach for sequence-specific knockdown of a target gene via mRNA degradation (Elbashir and others 2001). In the present study, we used 27-mer dsRNA, which can act as substrate for Dicer to generate mature 21-mer siRNA within the cell. These longer RNAs are processed by Dicer into 21-mer siRNAs in a predictable manner, with increased resistance to nuclease activity and increased potency, which is thought to arise from the participation of Dicer in RISC formation (Kim and others 2005).
The RNAi technology has been employed widely in the chick embryo to address questions related to developmental biology of the organism (Pekarik and others 2003; Dai and others 2005; Harpavat and Cepko 2006). There are at least 3 reports of the in vitro use of RNAi for downregulating expression of the chicken immune system genes involving iNOS, TLR3, and NF-κB (Cheeseman and others 2008; Karpala and others 2008; Chiang and others 2009). There has been no report of the utility of this technology to modulate expression of chicken cytokines, including IFN-γ.
Here, we report inhibitory activity of 3 siRNAs against IFN-γ using a cotransfection model system. The results presented here demonstrated that IFN-γ mRNA expression can be downregulated as early as 24 h and up to 72 h after treatment of cells with siRNA oligonucleotides. In addition to downregulation of IFN-γ transcripts, there was a reduction in the amount of IFN-γ protein present in the culture supernatant of IFN-γ-expressing cells that were transfected with siRNA. Generally, it is recommended (Wu and others 2004) that target protein levels be analyzed around 48–72 h post-transfection. In the present study, however, downregulation of IFN-γ protein was only significant at 24 and 48 h after treatment, consistent with our observations for IFN-γ transcripts. The slight discrepancy between the temporal pattern of protein downregulation in the present study compared to that of the previous studies may be related to the half-life of IFN-γ or the nature of the cell culture system we used. Here, we used cells transfected with a plasmid that constitutively expressed chicken IFN-γ. The CMV enhancer–promoter combination used in our IFN-γ expression plasmid is used for high-level and stable transgene expression (Ward and Stern 2002; Nitta and others 2005; Schlabach and others 2010). Therefore, IFN-γ expression under the influence of this promoter might well have exceeded the level of endogenous expression and the capacity of the siRNAs to silence it. Further, it has been demonstrated in several studies that siRNA effects are temporary. For example, an in vitro study examining the kinetics of siRNA gene silencing has reported mRNA and protein levels returning to the normal range 1–2 days after transfection (Bertrand and others 2002). In another study, the effects of siRNA remained for <1 week within dividing cell lines (Bartlett and Davis 2006). Our results were similar, with maximum knockdown that lasted 24–48 h, and thereafter diminishing.
An important question is the efficacy of selected siRNA to knockdown endogenous protein in a more biological relevant situation. To address this question, we stimulated primary splenocytes obtained from TROVAC-AIV H5-vaccinated birds using a known peptide (B7) to induce IFN-γ expression. Significant knockdown was observed in cells stimulated with the B7 peptide and transfected with siRNAs targeting chIFN-γ at both 12 and 24 h when compared with B7-stimulated cells.
One aspect of the siRNA technique that requires further attention is the off-target (or IFN pathway inducing) effects of these molecules. Studies have reported that treatment of cells with siRNAs may result in IFN-mediated activation of the Jak-STAT pathway. This activation is mediated by the dsRNA-dependent protein kinase PKR and OAS, which are induced by siRNAs (Sledz and others 2003). Therefore, the expression of 2 representative genes of the IFN pathway was examined to address whether there were any measurable off-target effects in cells treated with shRNA molecules. The studies presented here did not reveal upregulation of IFN-α (data not shown) and OAS genes, raising the strong possibility that the treatment did not have a major off-target effect. However, a recent report suggests that DF-1 cells may not be able to produce type-I IFN in response to 5′-triphosphate siRNA due to an absence of retinoic acid-inducible gene I protein in chickens (Barber and others 2010). However, in a subsequent study, a chicken MDA5 (ChMda5) homolog was identified and was shown to be involved in the IFN response of chicken cells to dsRNA (Karpala and others 2011).
In general, 2 different approaches can be employed for delivery of RNAi: synthetic RNA duplexes can be introduced directly into cells or alternatively; dsRNA can be expressed inside the cell using a plasmid or viral vector. The first direct delivery of siRNA in vivo was by administering short duplexes of naked siRNA (McCaffrey and others 2002). Some primary obstacles using this approach in vivo include uptake by nontarget cells, poor transfection efficiency, nuclease degradation, endosomal trapping, and excretion by the organism. To overcome these limitations, several groups have developed viral-based vectors to deliver shRNA into host cells (Bell and Brickell 1997; Tomar and others 2003; Bromberg-white and others 2004; Chen and others 2009). In the latter approach, RNAi effects can be sustained for a longer period of time.
Here, we report downregulation of chicken IFN-γ expression in cells using rAAAV in vitro. Earlier studies have utilized rAAAVs for the expression of a reporter gene in embryonic tissues in vitro, whereas other studies have shown the use of AAAV vectors for expression of viral proteins in vivo (Estevez and Villegas 2006; Perozo and others 2008a, 2008b; Wang and others 2009). Only a single recent study utilized rAAAV for expression of RNAi molecules. AAAV-based vectors are safe, and moreover, expression cassettes up to ∼4 kb in length can be packaged into AAV capsids without compromising infectivity. In our study, an expression cassette containing the chicken U6 promoter, and an shRNA was constructed and cloned into a plasmid vector containing the ITRs, followed by generation of rAAAV-expressing shRNA. Several groups have described the use of small nuclear RNA (snRNA) promoters (H1 or U6) for the expression of siRNA in chickens (Dai and others 2005; Kudo and Sutou 2005; Wise and others 2007). In this regard, we chose a polymerase III promoter (U6), because this promoter transcribes endogenous snRNAs and is most commonly used to express shRNA (Kudo and Sutou 2005; Bannister and others 2007). The mammalian pol III promoter, H1, can be used to transcribe shRNA in chicken cells (Yuan and others 2006); however, another study showed that the chicken U6 promoter was significantly better for expressing microRNA in chicken cells compared to promoters of mammalian origin (Das and others 2006).
In conclusion, the present in vitro study provides evidence that synthetic oligonucleotides (DsiRNAs) selected for IFN-γ were able to successfully reduce expression of this cytokine, without stimulating IFN-α or OAS expression. These findings provide a basis for future studies aimed at utilizing recombinant viral vectors to deliver genes or to knockdown expression of a gene of interest. These studies may be used to investigate the importance of IFN-γ and other cytokines in the chicken immune system as well as utilize the knowledge to improve vaccines.
Footnotes
Acknowledgments
This study was funded by the Ontario Ministry of Agriculture, Food and Rural Affairs, Poultry Industry Council, and the Natural Sciences and Engineering Research Council of Canada.
Author Disclosure Statement
No competing financial interests exist.