
Abstract
Interferon gamma (IFN-γ) is an important cytokine that induces antiviral, antiproliferative, and immunomodulatory effects on target cells, and is also crucial in the early defense against intracellular parasites, such as Listeria monocytogenes and Toxoplasma gondii. The biological activity of IFN-γ relies upon the formation of a complex with its 2 receptors, the interferon gamma alpha chain (IFNGR1) and beta chain (IFNGR2), which are type II cytokine receptors. Structural models of ligand–receptor interaction and complex structure of chicken IFNs with their receptors have remained elusive. Here we report the first structure of Gallus gallus (chicken) IFNGR1 (chIFNGR1) at 2.0 Å by molecule replacement according to the structure of selenomethionine substituted chIFNGR1. The structural comparison reveals its structural similarities with other class II cytokine receptors, despite divergent primary sequences. We further investigate the ligand–receptor interaction properties of chicken IFN-γ (chIFN-γ) and chIFNGR1 using size-exclusion chromatography and surface plasmon resonance techniques. These data aid in the understanding of the interaction of chicken (avian) IFN-γ with its receptors and its signal transduction.
Introduction
T
The Gallus gallus (chicken) interferon gamma (chIFN-γ) gene was cloned from a cDNA expression library generated from a T cell line and identified through its antiviral activity and immunoregulated macrophage activities (Digby and Lowenthal 1995; Song and others 1997). Chicken IFNGR1 (chIFNGR1) protein was generated and its physical characteristics were determined by mass spectrometry and circular dichroism (CD) spectrometry (Han and others 2006). Chicken IFNGR2 (chIFNGR2) was cloned using rapid amplification of cDNA 5′ and 3′ end (5′ and 3′ RACE) method and the secondary structure of its protein was identified by CD spectropolarimeter analysis (Han and others 2008).
Over the past decades, with the growth of biotechnology, such as CD spectroscopy, surface plasmon resonance (SPR), isothermal titration calorimetry, nuclear magnetic resonance (NMR), and X-ray diffraction, and others, it has become feasible to elucidate ligand–receptor interactions and structure properties of cytokines with their receptors. Human IFN-γ (huIFN-γ) binds IFNGR1 with high affinity (10−9–10−10 M) (Aguet and others 1988; Bach and others 1995; Walter and others 1995), and the crystal structure of the complex has been shown to have 2 molecules of IFNGR1 bind to the homodimer of IFN-γ forming a 2:1 complex, where the receptor molecules do not interact with one another and are separated by 27 Å (Walter and others 1995). Moreover, the crystal structure complex of glycosylated extracellular part of IFNGR1 bound to IFN-γ, in addition to the expected 2:1 complex, revealed the presence of a third receptor molecule not directly associated with the IFN-γ dimer, thereby forming 3:1 complex with its ligand IFN-γ (Thiel and others 2000). Recently, the structure of human IFN-λ1 complexed with human IFN-λR1 was determined at a 1:1 molar ratio (Miknis and others 2010). Further, the complex structure of human IL-10 with human IL-10R1 showed that the complex consisted of 2 IL-10s and 4 IL-10R1 molecules (Josephson and others 2001), and a putative IL-22 with human IL-22R1 complex was proposed based on the crystal structure of IL-22/IL-22R1/IL-10R2 complex model (Bleicher and others 2008). Therefore, the current data regarding the complexes formed between cytokines and their receptors suggests that the architecture of their interactions may govern the process of initial recognition of the signaling transduction pathway cascade.
So far, although there have been many studies on huIFN-γ and its receptors, there has been no report illustrating chIFN-γ/chIFNGR1 interaction and the structural basis of the ligand–receptor complex of chIFN-γ with its receptor remains elusive. Chicken, like fish and frog have not only become vertebrate models for the study of infectious diseases and tumors, but also, they are present at key positions in evolution and contribute significantly to studies related to development and comparative analysis of immune system function and development (Savan and others 2009). Consequently, it is meaningful to elucidate the interaction and structural properties between chIFN-γ and its receptors.
In the present study, we report the 2.0 and 2.5 Å X-ray crystal structure of selenomethionine (Se-Met) substituted and native chIFNGR1, respectively. Comparison of the high-resolution structure chIFNGR1 with those of other type II cytokine receptors showed it is highly conserved between chicken and human, although there are considerable differences in their primary amino acid sequences. To elucidate the interaction relationship between chIFN-γ and chIFNGR1, analytical size-exclusion chromatography and SPR techniques were used. We propose that a plausible model for the interaction of chicken (avian) IFN-γ with its receptors.
Materials and Methods
Cloning, protein expression, and purification
The chIFN-γ gene (GenBank accession No. AY163160.1) was amplified as described previously (Song and others 1997), C-terminal (residues 19–164) was subcloned into fusion vector pGEX-6p-1 (GE Healthcare) with an N-terminal GST-tag, a unique BamHI restriction site, a C-terminal stop codon, and a unique XhoI restriction site. The recombinant plasmids were transformed into Escherichia coli (E. coli) Rossetta-gami 2 (DE3) (Novagen), and expressed as soluble fusion proteins (GST chIFN-γ). Cells were harvested and lysed by an ultrasonic cell crusher with ice-cold phosphate buffer (pH 7.4), and then were centrifugated at 2,000g for 15 min at 4°C. The supernatant of lysate was applied into glutathione-Sepharose 4B (GE Healthcare) beads, after washing, soluble GST chIFN-γ was eluted by on-column cleavage with PreScission Protease (GE Healthcare) for 16 h at 4°C. The preliminary purified proteins were applied onto a Superdex 75 (GE Healthcare) size-exclusion column with ÄKTA purifier (GE Healthcare) for further purification.
ChIFNGR1 gene (GenBank accession No. NP_001123859.1) was constructed by our research group, previously (Han and others 2008; Ping and others 2012). The extracellular domain of chIFNGR1 (residues 28–234) was cloned into pET-21a vector (Novagen) with the restriction sites NdeI and XhoI, and a stop codon was used in C-terminal. Recombinant plasmids were transformed into E. coli strain Rossetta (DE3) and B834 (DE3) (Novagen) for the expression of native chIFNGR1 and Se-Met substituted chIFNGR1 (Se-Met chIFNGR1), respectively. Both of them were then expressed in inclusion bodies. The Se-Met chIFNGR1 was expressed using a modified methionine pathway inhibition procedure (Begley and others 2003). Transformed E. coli Rossetta (DE3) cells were grown at 37°C in 2×YT media (50 μg/mL ampicillin) to an optical density at 600 nm (OD600) about 0.6–0.8. Cells were harvested by centrifugation at 1,500g for 15 min and washed twice with M9 media [one liter of 10×M9 salts contains 60 g of disodium hydrogen phosphate, 30 g of monopotassium phosphate, 5 g of sodium chloride (NaCl), and 10 g of ammonium chloride; pH 7.4], and then resuspended in M9 media (50 μg/mL ampicillin) supplemented with 100 mg/L (Lys), 100 mg/L (Thr), 100 mg/L (Phe), 50 mg/L (Leu), 50 mg/L (Ile), and 50 mg/L (Val). Cell cultures were then grown for 15 min at 37°C to exhaust any residual methionine and 100 mg/L Se-Met was added. Isopropyl β-
For chIFNGR1 refolding, the procedure was done as described previously (Ping and others 2012). Briefly, cell pellets containing the inclusion bodies were harvested and washed three times with a buffer containing 0.5% v/v Triton X-100 [50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 10 mM ethylene diamine tetraacetic acid (EDTA), and 10 mM DL-dithiothreitol (DTT)], and then once with the same buffer without Triton X-100. The inclusion bodies were dissolved overnight in 6 M Gua-HCl [with 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 10 mM EDTA, 10% glycerol, and 10 mM DTT] using 1 mL of buffer per 30 mg inclusion bodies. The chIFNGR1 was refolded by the gradual dilution method using refolding buffer [100 mM Tris-HCl (pH 8.0), 400 mM
Crystallization, data collection, and processing of Se-Met chIFNGR1
The purified Se-Met chIFNGR1 was crystallized according to the native chIFNGR1 procedure (Ping and others 2012). The crystals of Se-Met chIFNGR1 were obtained in 20% (v/v) polyethylene glycol 5000 monomethyl ether, 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 0.5 M sodium carbonate (pH 7.0). For data collection, the crystals were cryoprotected in mother liquor containing 20% glycerol before being flash cooled directly in liquid nitrogen. Diffraction data were collected on beam line BL17U1 (100 K, wavelength 0.98 Å) of Shanghai Synchrotron Radiation Facility. The data were processed and scaled using the HKL2000 software package (Lake and others 1997).
Structure solution, refinement, and analysis
The crystal structure of Se-Met-IFNGR1 was firstly solved by the method of multiwavelength anomalous diffraction (Hendrickson and others 1990) by phase determining with Se-Met molecules. The structure of native chIFNGR1 was determined by molecular replacement method using Phaser (Read 2001) from the CCP4 program suite (Collaborative Computing Project 1994) using the structure of Se-Met chIFNGR1 as the model. The different residues between native chIFNGR1 and Se-Met chIFNGR1 were manually rebuilt in the program COOT under the guidance of F o–F c and 2F o–F c electron density maps (Emsley and Cowtan 2004). Consequently, the initial rigid body and a series of restrained translation/libration/screw (TLS) refinements were performed with the REFMAC5 program (Murshudov and others 1997). All of the structures were further refined by additional rounds of refinements using the PHENIX package (Adams and others 2002), with coordinate refinement, isotropic atomic displacement parameters (ADP) refinement, and bulk solvent modeling. The stereochemical quality of the final model was assessed with the PROCHECK program (Laskowski and others 1993). All structural figures were generated using PyMOL (DeLano 2002).
Mutagenesis of putative ligand binding sites of chIFNGR1
The chIFNGR1 mutations were subcloned using polymerase chain reaction (PCR) using recombinant plasmid chIFNGR1/pET-21a as previously report (Wei and others 2004), 2-step overlapping PCR was performed for 2 cycles using QuikChange® Site-Directed Mutagenesis Kit (Stratagene), and then DpnI (New England Biolabs) was added to the mixture to overdigest the methylated template for 90 min at 37°C. After that, 2.5 μL digestion product was transformed to Rossetta (DE3). All the mutations were expressed and purified according to the method of wild-type chIFNGR1, and the buffer of size-exclusion chromatographies was phosphate buffer (pH 7.4).
SPR analysis between chIFN-γ and chIFNGR1
SPR was performed using Biacore® 3000 (GE Healthcare) to determine the interaction of chIFN-γ and chIFNGR1 (Cooper 2003; Sitlani and others 2007). Purified chIFNGR1 was coupled to a research-grade carboxymethylated dextran sensor chip (CM5; GE Healthcare) to give surface densities of ∼1,500 resonance units (RU). ChIFN-γ proteins were injected with the concentration of 0, 15.625, 31.25, 62.5, 125, and 250 nM in a solution of 10 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% surfactant P20. The surfaces of sensor chip were regenerated with 15 mM sodium hydroxide and all of the injections were performed at 25°C with the flow rates of 5 μL/min. The data of measurements were recorded with real-time and analyzed by BIAevaluation software version 4.1, and then fit by assuming a simple 1:1 Langmuir binding model.
For the determination the interaction of chIFNGR1 mutations with chIFN-γ, the chIFN-γ (residues 19–164) was subcloned into pET-28a vector (Novagen) with the N-terminal His-tag, which was purified by Ni-NTA column (Novagen) and washed by gradient imidazole, the Superdex 75 (GE Healthcare) size-exclusion column with phosphate buffer (pH 7.4) was used for further purification. SPR analyses were still performed by using Biacore 3000 equipped with Sensor Chip CM5. First, His4 mAb surfaces were prepared by using standard procedures (Rich and others 2002), ∼13,000 RU antibody immobilized on the chip. Second, the ligand chIFN-γ/His (20 μg/mL) was injected across immobilized His antibody, resulting in the capture of ∼1,700 RU ligand. Third, 1 μM of each mutation of chIFNGR1 and the wild type were injected over antibody-captured chIFN-γ/His. The ligand/receptor complexes were stripped for regeneration with elution buffer: 10 mM phosphoric acid. The binding status was recorded and analyzed by BIAevaluation software version 4.1.
Results
Overall structure of chIFNGR1
Due to the limited sequence identities of chIFNGR1 with IFN receptors of known structures, our initial molecular replacement trials failed. The presence of 3 methionine in the protein sequence reminds us that the Se-Met-based single wavelength anomalous diffraction (SAD) method could be used to solve the phase problem. Finally, all the 3 methionine sites were successfully located, based on which a model of good stereochemistry was built. The final structure was refined to R work =0.2334 and R free =0.2602, respectively (Table 1). Single crystals of native and Se-Met chIFNGR1 were successfully obtained, which could diffract to 2.5 and 2.0 Å resolution, respectively (Ping and others 2012). The solved structure, with 207 residues being visible in the electron density map, contains 2 Ig-like domains (D1 and D2) connected by a flexible linker (residues 126–131) (Fig. 1). The D1 domain is composed of N-terminal residues 28–125 and forms a β sandwich structure with a layer of 5 β-strands. In contrast to domain D1, the D2 domain of chIFNGR1, which includes 103 amino acids from 132 to 234, forms a 7-stranded β sandwich structure. There are 3 disulfides: C51&C59 in D1 domain, C162&C167, and C181&C202 in D2 domain (Fig. 1). Three methionine residues, one located in the D1 domain remaining two located in the D2 domain, were substituted with Se-Met to help determine the phase of the chIFNGR1 structure. The structure of chIFNGR1 reveals that this protein is largely formed by β-strands, with only 13 residues forming 2 α-helices (Fig. 1).

The ribbon diagram of chIFNGR1 structure. An overall view the crystal structure of chIFNGR1 displays as 2 domains: D1 and D2, which contain 5 and 7 yellow-colored β-sheets, respectively. Helices are colored in red. Three disulfide bridges link counterpart molecules are indicated as blue sticks. chIFNGR1, Gallus gallus (chicken) IFNGR1; IFN, interferon.
Values in parentheses are for the highest-resolution shell.
PDB ID, protein data bank identification.
Comparison of the 3-dimensional structure of chIFNGR1 with other cytokine receptors
Alignment of primary sequences reveals that chIFNGR1 has lower identity with other class II cytokine receptors, including huIFNGR1, huIL-10R1, and huIL-22R1 (Fig. 2). Nevertheless, the 3-dimensional structures of these proteins are quite similar with each other. Structural comparison of chIFNGR1 with huIFNGR1 reveals that chIFNGR1 almost shares the similar structure with its human counterpart (Fig. 3), although their sequence identity shows just about 30% (Fig. 2). The structure comparison onto those of huIFNGR1, huIL-10R1 and huIL-22R1, showed they almost share the same structure with chIFNGR1 (Fig. 3). With the root-mean-square deviation (RMSD) was varied according to the comparison of chIFNGR1 with other cytokine receptors (Table 2). However, a remarkable additional α-helix located in D2 domain was detected in chIFNGR1, which was absent in other counterparts, and the 2F o–F c electron density map of which was shown in Fig. 4.

Multiple sequence alignment of select members of the type II cytokine receptors family, including chIFNGR1, huIFNGR1, huIL-10R1, and huIL-22R1. The numbering used for huIFNGR1, huIL-10R1, and huIL-22R1 is aligned on the numbering of the sequence of chIFNGR1. The secondary structures are indicated for each sortase with black arrows (β-sheets) and black coil (α-helices). The similar sequence of chIFNGR1 and huIFNGR1 was shown with red square. The red shading indicates the region that is conserved in all compared sequences, and the blue square indicates the identity >50% in all sequences. Three disulfides bonds in the chIFNGR1 molecule were labeled in the following of their correspondence sequence. The residues marked in red indicate the binding sites of huIFNGR1 with its ligand huIFN-γ. The sequences were aligned using ClustalW2 (
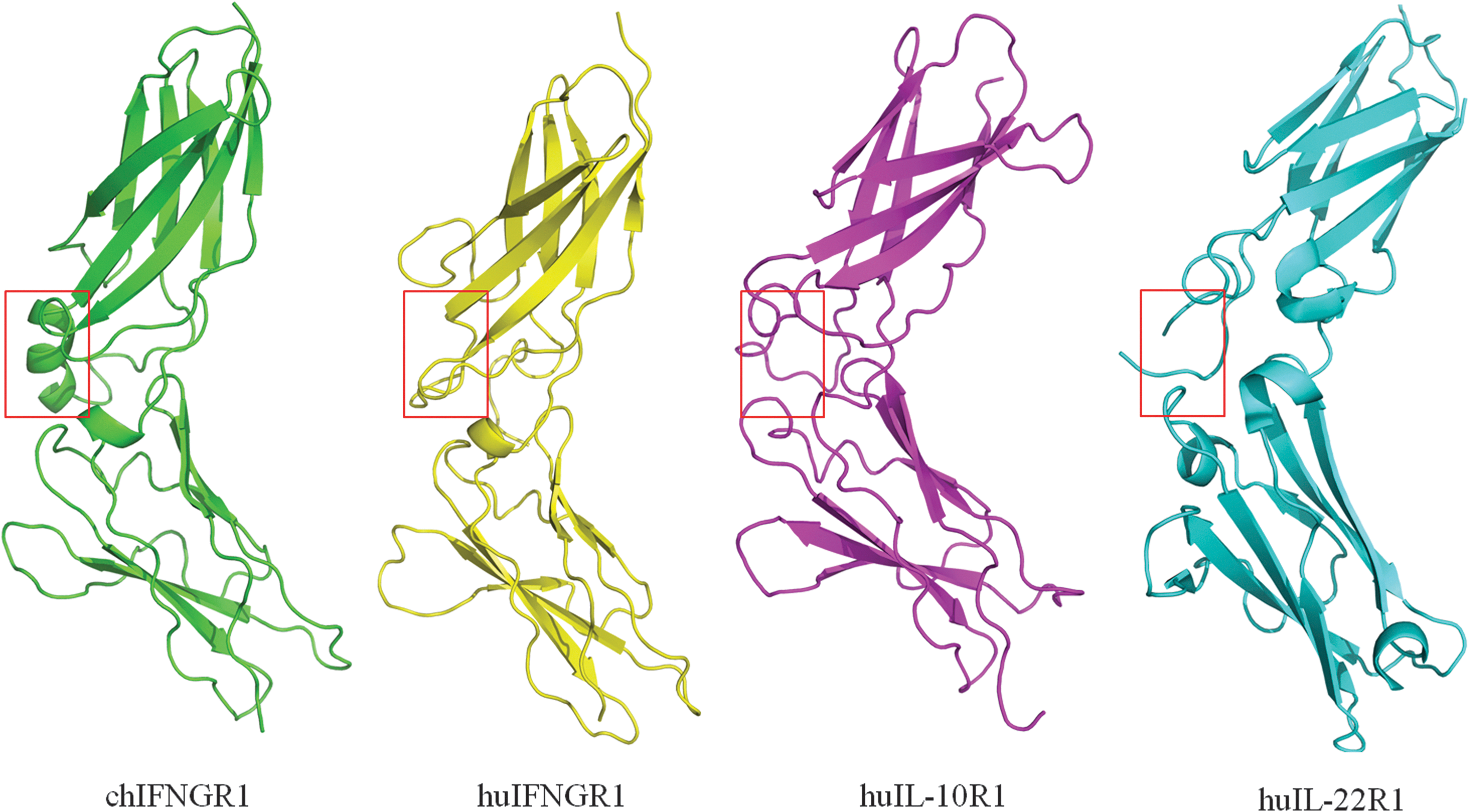
The structures of type II cytokine related receptors. Structural comparison of the 4 type II cytokine related receptors, chIFNGR1, huIFNGR1, huIL-10R1, and huIL-22R1. The structures of chIFNGR1 (green), huIFNGR1 (yellow), huIL-10R1 (magenta), and huIL-22R1 (cyan) are superimposed. The ribbon structures were displayed with PyMOL (DeLano Scientific;
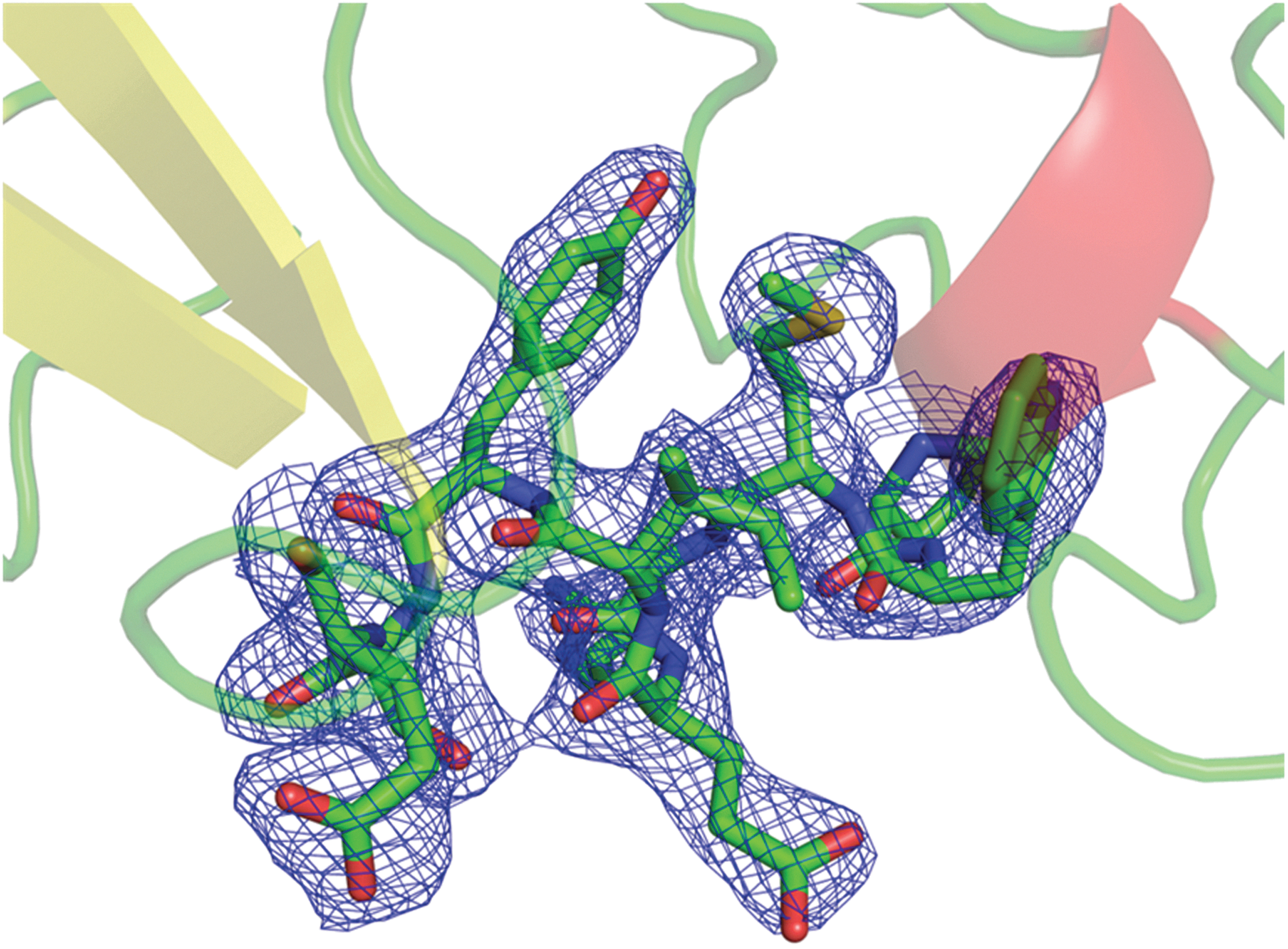
The 2F o–F c electron density map of the α2-helix of chIFNGR1 structure which is absent in the other type II cytokine receptors.
RMSD, root-mean-square deviation.
Structural alignment of this newly obtained chIFNGR1 structure with the huIFNGR1 reveals the well-aligned Ig-like domain, but relative differences lies in the interdomain angles between them. As shown in Fig. 5, with the longitudinal axis crossing the first Ig domain of huIFNGR1 as reference, the orientation of the chIFNGR1 structure could be rotated about 30.9°. This observation indicates that the relatively flexible interdomain linker for the 2 domains in the IFNGR1 structure, which is similar with the PD-L1 structure (Chen and others 2010).
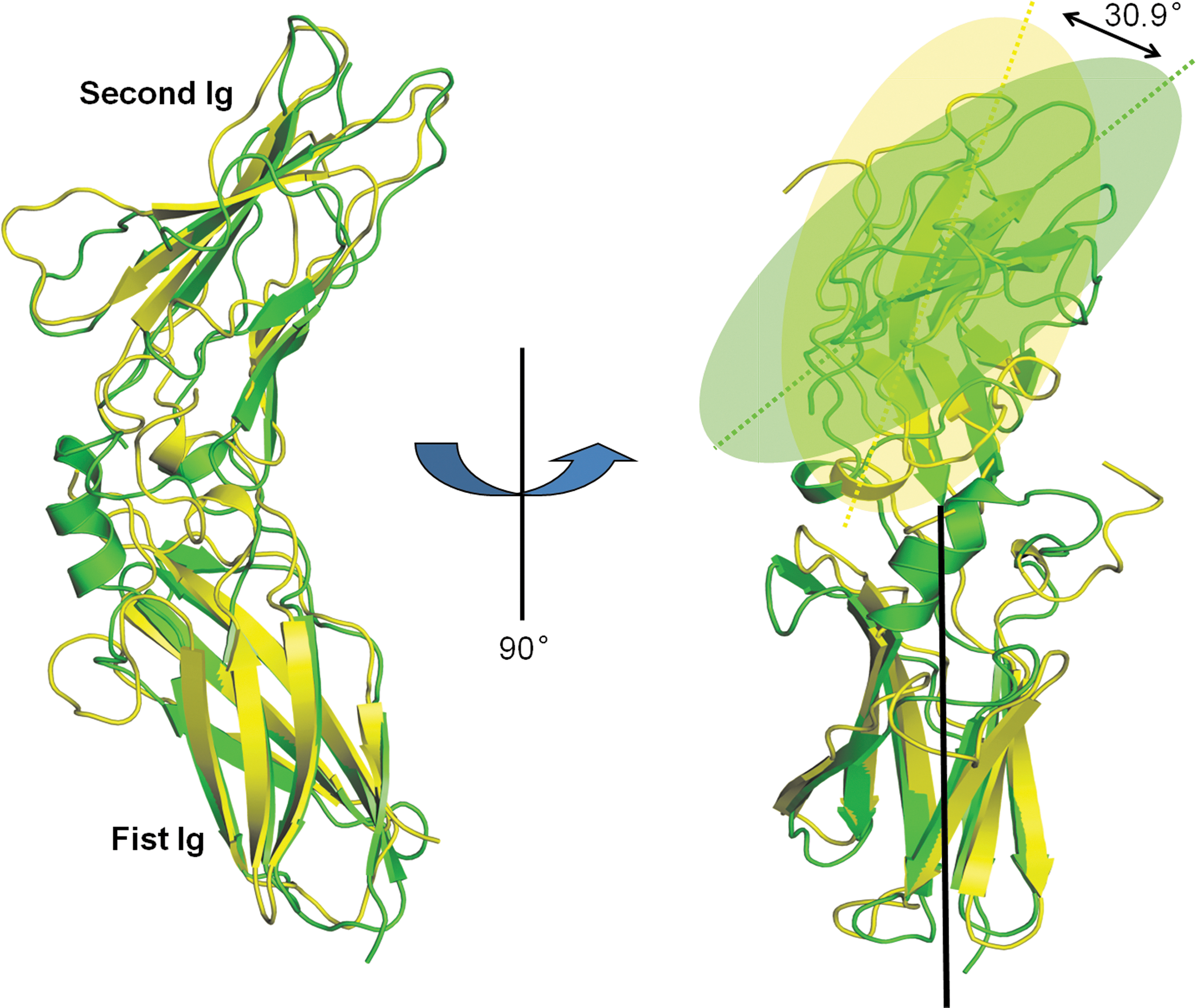
The superimposition of chIFNGR1 with huIFNGR1 structures. The 2 structures of free and unbounded form of chIFNGR1 solved in this study colored in green, and the huIFNGR1 colored in yellow. Their overall structure is quite similar (shown on the left side). After rotation with an angle of 90°, with their first Ig domains superimposed, the second Ig domains are related by a 30.9° rotation. The colored dash lines represent the second Ig domains within the huIFNGR1 and chIFNGR1, respectively (seen in the right side).
The complex structure of human IFN binding to its receptor IFNGR1 has been reported by Thiel and others (2000) previously. In this structure, 12 residues in huIFNGR1 are identified as the key players involved in the ligand–receptor interaction. Most of these residues are located in the loop regions, which include “the β3-β4 loop” and “the β4-β5 loop” in D1 domain, and “the β2-β3 loop,” “the β4-β5 loop,” and “the β6-β7 loop” in D2 domain. For this ligand-binding related loop, chIFNGR1 exhibit a quite similar loop as that of the huIFNGR1. Twelve ligand binding sites were found by analyzing the complex structure of huIFNGR1 with huIFN-γ (Fig. 6), which were gathered together in the loop region. chIFNGR1 has similar loop region to huIFNGR1, which contains the majority of binding sties K38A, L41A, S42A, K44A, H70A, W73A, E89A, E92A, and F189A, which help chIFNGR1 anchoring to its ligand huIFN-γ. However, 3 other binding sites T158A, V159A, and D191A, show a large conformational change in chIFNGR1 and huIFNGR1 structures (Fig. 6). The conformational change may occur to allow for the interaction of amino acids between chIFNGR1 and chIFN-γ in the binding site, although the similar region between chIFNGR1 and huIFNGR1 in the binding site does not show this configuration.
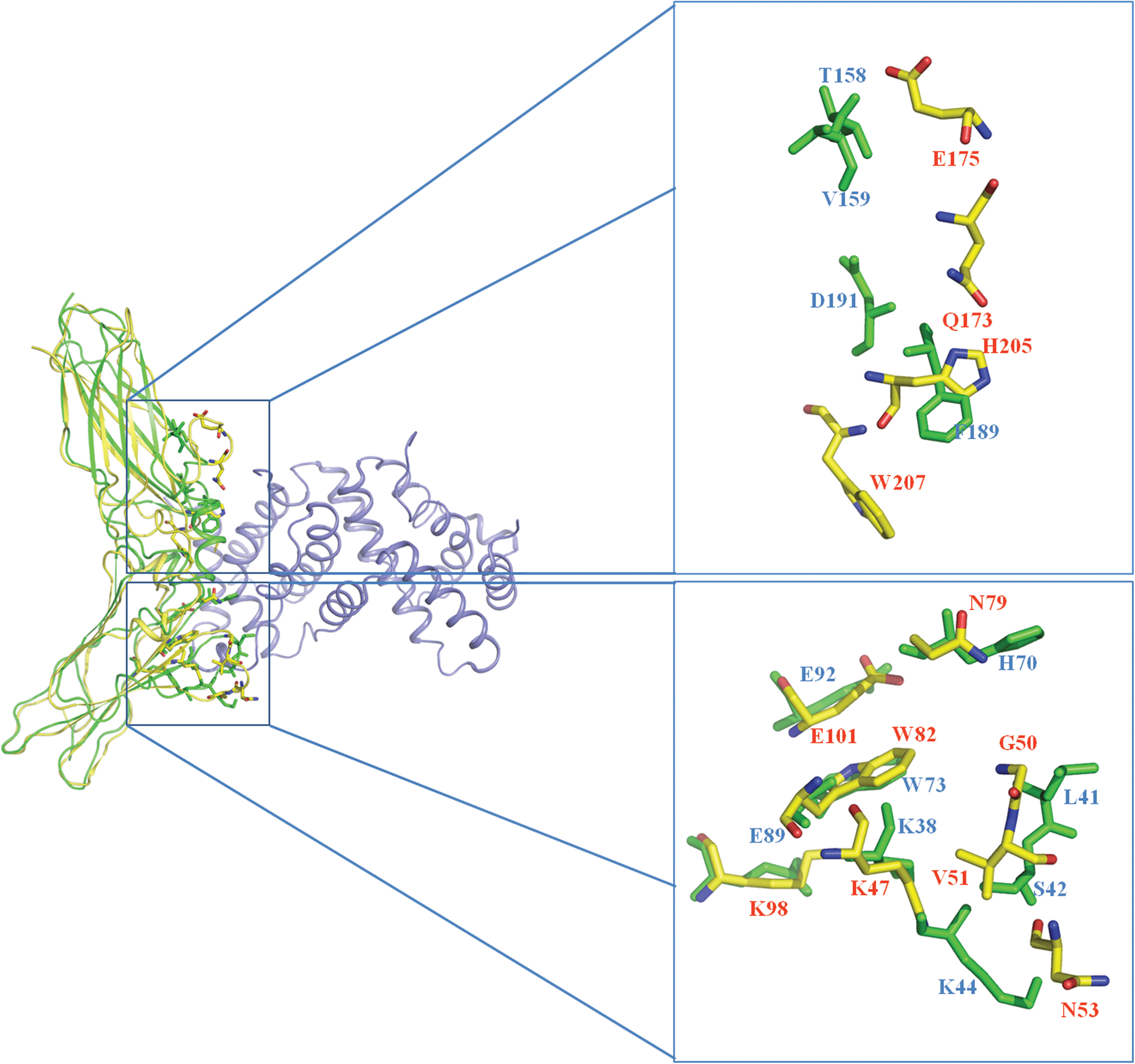
Close-up view of the ligand–receptor binding sites comparison between chIFNGR1 and huIFNGR1. The binding sites of huIFN-γ &huIFNGR1 complex were analyzed and marked with yellow sticks, and the green ones represent the counterpart sites in chIFNGR1. chIFNGR1 (green), huIFNGR1 (yellow), huIFN-γ (blue).
Interaction characterization of soluble chIFN-γ with chIFNGR1
Without a complex structure of chIFN-γ binding to chIFNGR1, we cannot define the detailed residue-by-residue interaction mediating intimate interaction between the 2 molecules. Nevertheless, the sequence alignment result shows that 9 (out of 12) amino acids involved in ligand binding in huIFNGR1 are also conserved in the chicken receptor.
A 1:1 binding mode of chIFN-γ with chIFNGR1 was obtained using analytical size-exclusion chromatography with Superdex®75 10/300 GL column (GE Healthcare). As shown in Fig. 7, purified chIFN-γ and chIFNGR1 were eluted as a single peak at 12.96 and 10.99 mL, corresponding to an apparent molecular weight (MW ) of about 17 and 24 kDa, respectively. This result demonstrates that both chIFN-γ and chIFNGR1 exist as a homogeneous monomer in solution. In vitro mixture of chIFN-γ with chIFNGR1 yield a stable complex, which behaves as a single peak with an elution volume of about 10.08 mL. The molecular weight of the complex was calculated to be about 40 kDa, which is in good accordance with the MW of one chIFN-γ molecule in combination with a single chIFNGR1. We further demonstrated the identity of the complex by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 7).

The elution profiles of chIFN-γ, chIFNGR1, chIFN-γ and chIFNGR1 complex were displayed by size-exclusion chromatograph, which was carried out with a Superdex™ 75 10/300 GL column (GE Healthcare). The molecular weight of elution volume was marked according to the instructions of the column, and elution volume of every peak was labeled on the top of them. 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) results showed that the correct molecular weight of chIFN-γ and chIFNGR1 monomers, and they could bind with each other with 1:1 ratio. The molecular weight of chIFN-γ and chIFNGR1 are 17 and 24 kDa, respectively, which are correspondent with their elution volume of size-exclusion chromatography.
We also applied SPR using Biacore 3000 to detect the binding affinity of chIFN-γ with chIFNGR1. SPR is an optical technique that has gained wide recognition as a valuable tool to investigate biological interactions. SPR offers real-time in situ analysis of dynamic surface events and thus, is capable of defining the rates of both adsorption and desorption for a range of surface interactions (Green and others 2000). First, chIFNGR1 was immobilized onto CM5 sensor surface with ∼1,500 RU. Then gradient concentration (0, 31.25, 62.5, 125, and 250 nM) of chIFN-γ was injected, and the data were real-time recorded (Fig. 8). ChIFN-γ exhibited dramatically high binding affinity (K D=2.3×10−8 M) to chIFNGR1, and the kinetic data (k on was 1.25×105 M−1·s−1 and k off was 2.68×10−3 s−1) fit well with the 1:1 binding model, which was consistent with the size-exclusion chromatography assay. The putative binding sites of chIFNGR1 to chIFN-γ were prepared using site-directed mutagenesis method, and purified like the wild-type chIFNGR1 (Fig. 9a). The analysis result of the binding property of those mutations to chIFN-γ using SPR indicated that the K38A, L41A, H70A, E92A, F189A, and D191A mutations had no binding capability to chIFN-γ; however, the other mutations S42A, K44A, W73A, E89A, T158A, and V159A still could bind to chIFN-γ (Fig. 9b), the binding parameters was shown in Table 3.
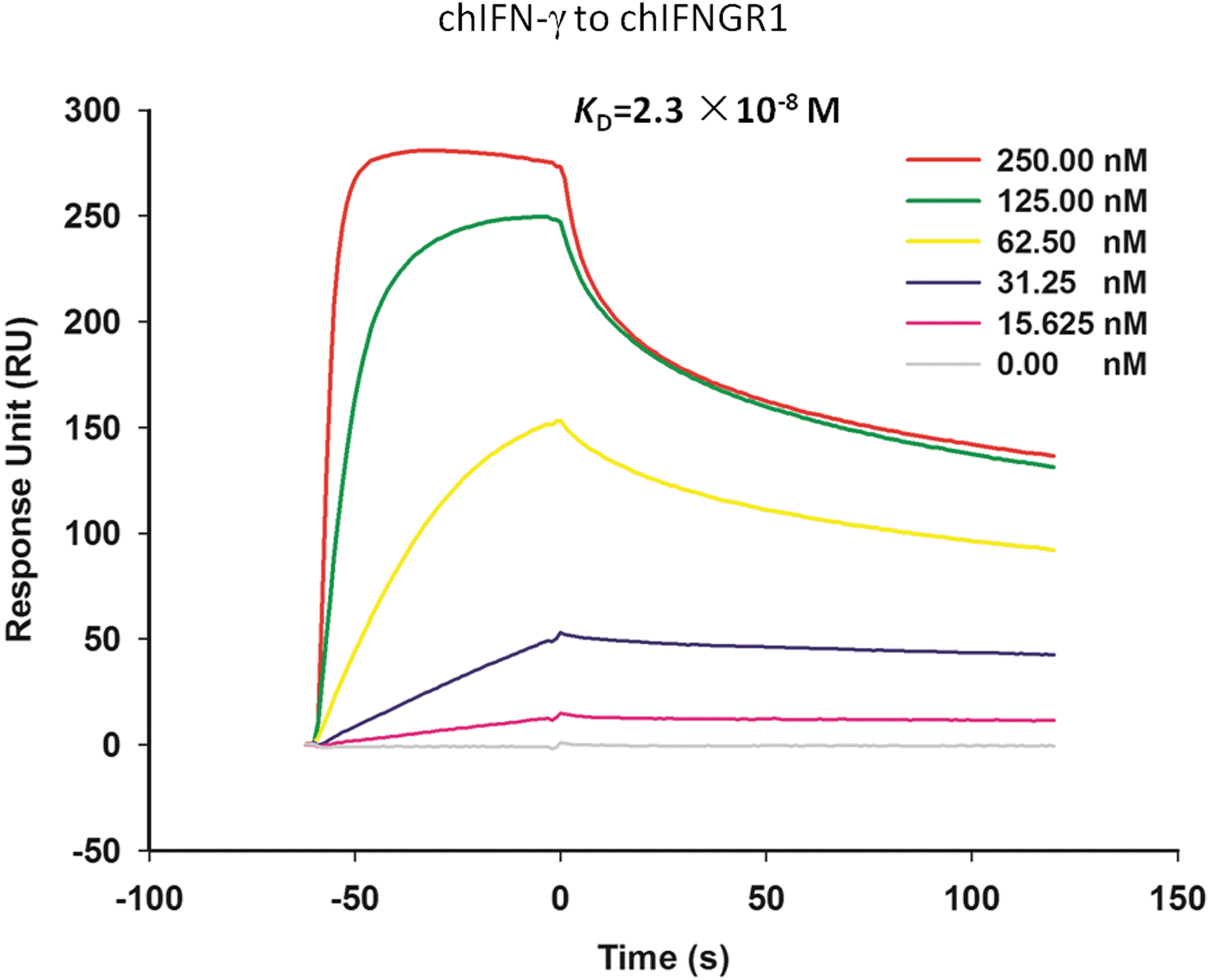
Surface plasmon resonance measurement of chIFN-γ monomer binding to immobilized chIFNGR1 monomer. chIFNGR1 was immobilized on the CM5 sensor chip with ∼1,500 RU. chIFN-γ protein was injected as mobile phase with the concentration of 15.625, 31.25, 62.50, 125.00, and 250.00 nM. All injections were performed at 25°C and measured using Biacore 3000 instrument (GE Healthcare). Kinetics analysis was carried out using the BIAevaluation software version 4.1, and then fit by assuming a simple 1:1 Langmuir binding model. The K D of chIFNGR1 to chIFN-γ was 2.3×10−8 M.
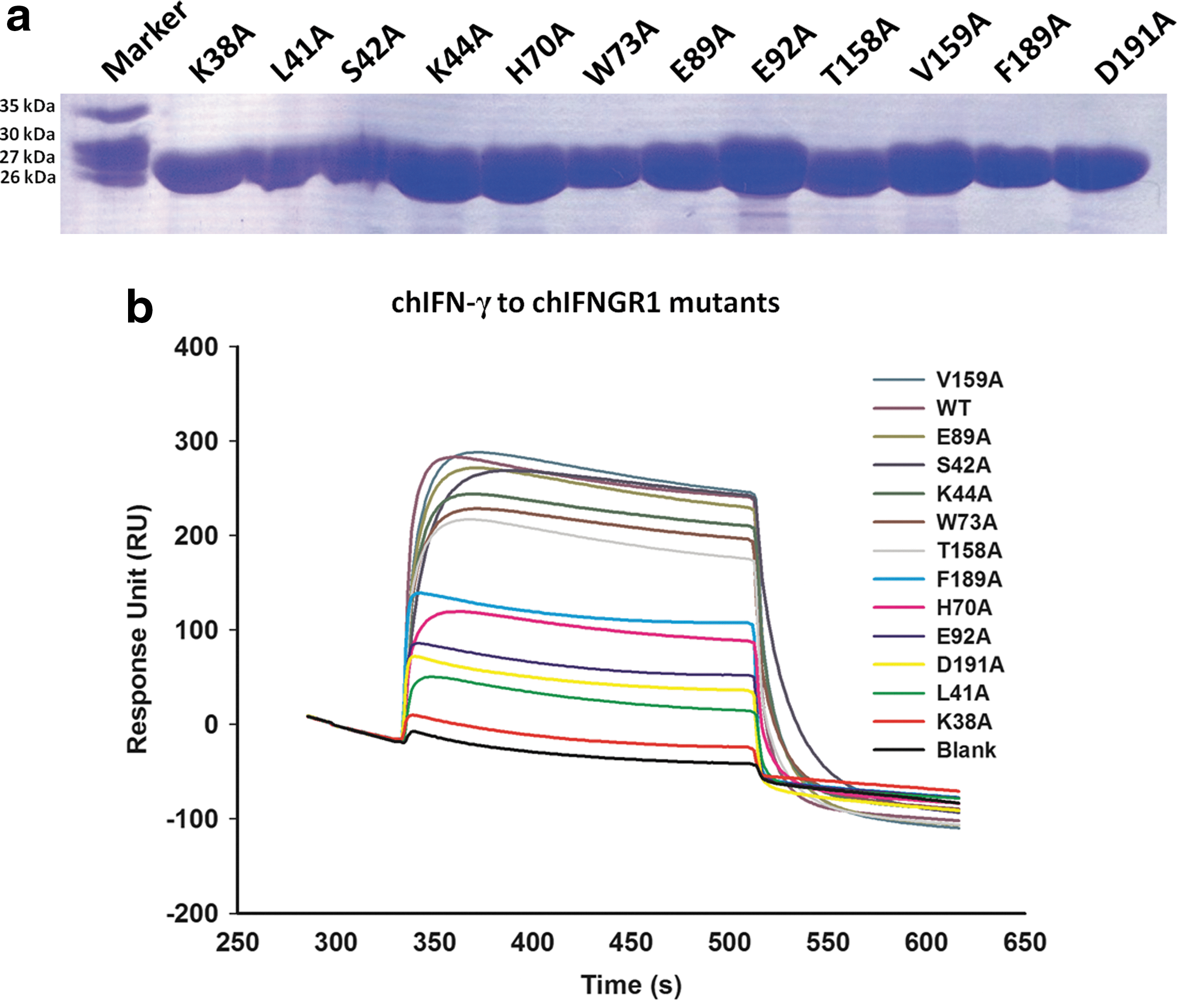
The purification of chIFNGR1 mutations and their binding capacities to chIFN-γ.
Discussion
The activation of numerous cytokines relies on ligand–receptor association, which is responsible for the initiation of signaling transduction pathway cascades (Platanias 2005; Sadler and Williams 2008). Here we expressed the recombinant chIFN-γ and one of its receptor chIFNGR1 in prokaryotic cell (E. coli) as soluble proteins and inclusion bodies, respectively.
In contrast to the dimerization IFN-γ of other species (Ealick and others 1991; Samudzi and others 1991; Randal and Kossiakoff 2000), chicken IFN-γ form monomers according to the elusion volume of size-exclusion chromatography (Fig. 7), and binds to chIFNGR1 with 1:1 ratio, which was confirmed by the Biacore 3000 SPR and possessed high binding affinity (K D=2.3×10−8 M), approaching to the ligand–receptor binding affinity (K D =10−9–10−10 M) of huIFN-γ to huIFNGR1 (Marsters and others 1995; Walter and others 1995). Moreover, further investigation using size-exclusion chromatography supported the result of SPR. All of this information suggests that chIFN-γ, like huIFN-γ, could bind to the alpha chain of its receptor (chIFNGR1) with high affinity with a 1:1 mode. However, the diversity of species results in the difference form of the configuration of their ligand: monomerization to chIFN-γ and the dimerization of huIFN-γ.
Interestingly, although there is only low sequence conservation between chIFNGR1 with other class II cytokine receptors, such as human IFNGR1 (huIFNGR1), human IL-10R1 (huIL-10R1), and human IL-22R1 (huIL-22R1) the structural comparison demonstrated that the chIFNGR1 shares tremendous similarity with other cytokine receptors. After superimposing the chIFNGR1 to huIFNGR1& huIFN-γ complex, the superimposed majority binding sites of the complex provided evidence for the chIFNGR1 binding sites in the structural level. It could be inferred that the binding sites of chIFNGR1 to its ligand could be focused on those sites, although there is no structure solved for the chIFNGR1& chIFN-γ complex.
In conclusion, the monomer of chIFN-γ and chIFNGR1 could bind each other with high affinity and a 1:1 binding mode. Moreover, the structural superimposition of chIFNGR1 with the single crystal structure of other class II cytokine receptors and human IFN-γ&IFNGR1 complex indicates the similar structure of chicken chIFNGR1 and other cytokine receptors. Further, chIFNGR1 shares the same binding sites with the huIFNGR1 to chIFN-γ, which provides structural evidence for chIFNGR1 binding to its ligand chIFN-γ in a similar manner.
Footnotes
Acknowledgments
We thank the beamline scientists at Shanghai Synchrotron Radiation Facility (SSRF-beamline 17U) for technical support during data collection and Dr. Zheng Fan for assistance with SPR experiment. This work was supported by National Natural Science Foundation of China (NSFC; grant no. 31072118) and National Natural Science Foundation of China (NSFC; grant no. 81021003). Coordinates and structure factors are deposited in the Protein Data Bank (PDB code 4EQ2 for native chIFNGR1 and 4EQ3 for Se-Met chIFNGR1).
Author Disclosure Statement
No competing financial interests exist.