
Abstract
The mucosal surface of the gastrointestinal tract directly interacts with the mucosal lumen, which is continuously exposed to foreign antigens. Specialized intraepithelial lymphocytes (IELs), located between the basolateral surfaces of the epithelial cells, are important as the first line of defense against microbes as well as for their role in the maintenance of epithelial barrier homeostasis. Although IELs are mainly composed of T cells, they are phenotypically and functionally distinct from T cells in peripheral blood or the spleen. Not only are IELs stimulated by the antigens of the intestinal lumen but are they also stimulated by regulatory immune cells. The integrity of the intestinal mucosal barrier is closely tied to the IEL function. Cytokines produced by IELs modulate the cellular functions that trigger the downstream signaling pathways and mediate the barrier homeostasis. In this review, we will address the broad spectrum of cytokines that are derived from IELs and the functional regulation of these cytokines on the intestinal barrier.
Introduction
T
There is a group of cells at the basolateral side of intestinal epithelial cells (IECs) known as intraepithelial lymphocytes (IELs). These cells are in direct contact with the enterocytes and are in immediate proximity to antigens in the gut lumen. Although primarily comprised of CD8+ T cells, IELs are a heterogeneous population of T cells. IELs are classified into 2 major subgroups based on the phenotypic and functional characteristics (Hayday and others 2001). We will discuss these 2 groups of cells below. Not only do these cells sense pathogens in the gut but they also maintain homeostasis of the intestinal mucosal barrier (Yang and others 2005; Saurer and Mueller 2009). However, in addition to homeostatic functions, these IELs can also contribute to immune pathology and inflammatory diseases.
Although IELs undoubtedly possess a variety of functions, it is clear that these cells have a great impact on the intestinal barrier (Kunisawa and others 2007). In some physiological and pathophysiological conditions, IEL cytokine expression is altered, and these changes could mediate the observed physiologic changes in the intestinal epithelium (Yang and others 2002). In this review, we will discuss how adaptable is the mucosal barrier, and can be regulated in response to the cytokines produced by IELs.
Anatomy of Mucosal Barriers
Mucin as a major EC surface barrier
The gut epithelium is covered by a thick mucus layer that prevents direct contact of the epithelium with microorganisms and protects the IECs from sheer forces and other physical trauma from particles within the lumen (Hansson and Johansson 2010). The mucus layer is largely composed of mucin, which contains various digestive enzymes, antimicrobial peptides, and immunoglobulins (McGuckin and others 2011). Specialized ECs secrete mucins, and ECs and Paneth cells secrete antimicrobial peptides (such as cathelicidins and defensins), and plasma cells secrete immunoglobulin A (IgA). The mucus gel provides a matrix for the retention of antimicrobial molecules, which bind, aggregate, or destroy bacteria to prevent their adherence and subsequent translocation to the submucosa (Macpherson and others 2008; Bevins and Salzman 2011). Defective mucus layers result in increased bacterial adhesion to the surface epithelium, increased intestinal permeability, and enhanced susceptibility to dextran sodium sulfate (DSS)-induced colitis (Kim and Ho 2010).
Cellular components of the intestinal barrier
There is a complex array of cell types within the epithelium, including absorptive enterocytes, mucus-producing goblet cells, enteroendocrine cells and Paneth cells. IECs also contribute to mucosal immune surveillance through the expression of a wide range of pattern recognition receptors, such as Toll-like receptors (TLRs) and intracellular nucleotide-binding oligomerization domain-like receptors (Marques and Boneca 2011). Furthermore, a complex crosstalk between IECs and other players in the immune system, including T and B lymphocytes, IgA-secreting plasma cells, mast cells, dendritic cells, and macrophages, maintains a balanced immune response against intestinal antigens aimed at preserving gut homeostasis.
The intercellular complex
In addition to an intact EC layer, the paracellular pathway between cells must be sealed. The intercellular complex, composed of TJs, adherens junctions, desmosomes (DMs), and gap junctions, maintains this barrier. TJs and subjacent adherens junctions, which consist of transmembrane and cytoplasmic scaffolding proteins supported by a dense perijunctional ring of actin and myosin, form the apical junctional complex (AJC) and regulate the epithelial paracellular permeability (Gumbiner 1996; Popoff and Geny 2009).
TJs are located at the luminal side of the epithelium and act to limit the paracellular transport of ions and the translocation of luminal antigens (microorganisms and their toxins). They exhibit a complex molecular architecture involving the regulated interaction of cytoplasmic adaptor proteins [eg, zonula occludens (ZO)] and integral membrane linker proteins (eg, occludins and claudins) (Ashida and others 2012). The claudin family is the most important transmembrane protein member, and the tissue-specific subset of claudins determines the barrier properties of the respective epithelium (Zeissig and others 2007). Among 24 claudins, claudins 1, 4, 5, 7, 8, and 14 increase epithelial resistance predominantly by decreasing cation permeability, while claudins 2 and 16 counteract this resistance by creating cation-selective pores (Van Itallie and Anderson 2006). The different expression levels and distributions of claudins may allow for specific physiological functions along the length of the intestine. However, the role of occludin, a transmembrane TJ protein that interacts directly with claudins and actin, is less understood. Furuse and others found that occludin localized to the epithelial and endothelial TJs and was subsequently confirmed as the first integral membrane TJ protein (Furuse and others 1993; Cummins 2012). However, its function has not yet been fully delineated. To our surprise, TJ assembly and paracellular permeability remained unchanged in occludin knockout animals; the investigators noted that occludin likely played a larger role in TJ stability and barrier function than in TJ assembly (Saitou and others 2000). Sakakibara and others found that occludin was highly phosphorylated on Ser and Thr residues in the resting epithelium (Sakakibara and others 1997; Seth and others 2007), but very low levels of Tyr-phosphorylated occludin were present (Rao and others 2002). During the disruption of TJs by various factors, occludin undergoes dephosphorylation of Ser/Thr residues and increases phosphorylation of Tyr residues (Rao 2009). These transmembrane proteins are connected to the actomyosin cytoskeleton filaments through scaffolding proteins (ZO-1, ZO-2, and ZO-3), and together with cingulin, ZOs play a crucial role in TJ assembly and maintenance. ZO-1, 2, and 3 contain 3 PDZ domains (PDZ1–3), 1 SH3 domain, and 1 GUK domain (Itoh and others 1993; Beatch and others 1996; Haskins and others 1998), indicating that they belong to the TJ membrane-associated guanylate kinase-like homolog (MAGUK) family (Anderson and others 1995). ZO-1 and ZO-2 knockout mice reportedly exhibit embryonic lethality, while ZO-3 knockout mice lack an obvious phenotype (Katsuno and others 2008; Xu and others 2008).
Adherens junctions are the other members of the apical junction, which is immediately subjacent to the TJ and requires the activity of lineage-specific Ca2+-dependent adhesion proteins. These adhesion proteins, termed as cadherins, provide the strong adhesive bonds that maintain cellular proximity and are also a site of intercellular communication (Grunwald 1993; Alattia and others 1999). E-cadherin is functionally linked to the generation of a polarized epithelium (Wheelock and Jensen 1992). The ectodomain of E-cadherin extends from the cell surface to mediate the cell–cell contact, whereas its cytoplasmic tail contains the binding sites to interact with catenins and other regulatory proteins (Perez-Moreno and Fuchs 2006; Shapiro and Weis 2009). The basal levels of cadherin internalization would be expected to support their metabolic turnover and perhaps contribute to local remodeling of cellular contacts (Yap and others 2007). In addition to its role as a structural molecule, the cadherin–catenin complex is also involved in several key signal transduction networks. The structural integrity of the cadherin–β-catenin complex is determined by its phosphorylation status. Phosphorylation of E-cadherin or β-catenin by the Ser/Thr kinase CK II modulates the affinity between these 2 molecules, ultimately modifying the strength of cell–cell adhesion (Bek and Kemler 2002).
Lastly, subjacent to the AJC are spot-like intercellular junctions, referred to as DMs, which are located beneath the AJC. Each desmosomal plaque consists of a thick outer dense plaque and a translucent inner dense plaque (Thomason and others 2010). Desmogleins (DSGs) and desmocollins (DSCs), which belong to the desmosomal cadherin family and form homodimers and heterodimers, constitute the extracellular link between cells and are responsible for the appearance of the dense midline. The intracellular domain of DSGs and DSCs binds the Armadillo family proteins plakoglobin (PG) and the plakophilins, which in turn tether DMs to the intermediate-filament network (Brooke and others 2012).
IELs Are Unique and Heterogeneous
IELs are comprised of heterogeneous populations with varying phenotypes and immunological functions. IELs are predominantly CD8+ T cells and are immunologically distinct from peripheral T cells (Kunisawa and others 2007). IELs constitutively express CD103 (αE integrin), and they often express CD8αα homodimers, particularly in the small intestine, which are barely detectable in systemic immune compartments (Cepek and others 1994). Unlike T cells in other peripheral immune compartments, small and large IELs bear some, but not all, of the properties of activated T cells and are therefore partially activated. Upon isolation from the intestinal epithelium, TCRαβ+ and TCRγδ+ IELs are cytolytic in the absence of stimulation, and most IELs express markers of activated T cells, such as the CT antigen (in mice), CD69, or CD11c, the latter being a marker of activated intestinal CD8+ T cells (Viney and others 1990; Montufar-Solis and others 2007). However, they do not express CD25 or CD122, which are present on activated T cells. Furthermore, IELs are made up by a larger proportion of TCRγδ+ cells than what is found in the circulation and can constitute up to 60% of small-intestinal IELs (Bonneville and others 1988). IELs can be divided into 2 major subsets based on these unique features, which will be discussed in the following section.
Type-a IELS
Type-a IELs express αβTCRs with CD4 or CD8α and arise from conventional T cells, which are major histocompatibility complex (MHC) class II- and MHC class I-restricted, respectively. In contrast to type-b IELs, type-a IELs undergo conventional thymic selection and differentiate into naïve CD4+ and CD8αβ+ TCRαβ+ T cells that migrate to the periphery. These naïve T cells can differentiate into effector T cells in response to peripheral antigens, and they subsequently migrate to the gut and become incorporated into the induced IEL compartment (Cheroutre and others 2011). They express several markers, such as CD2, CD5, CD28, cytotoxic T-lymphocyte antigen-4, and Thy-1 (Klein 1986; Lefrancois 1991; Ohteki and MacDonald 1993; Van Houten and others 1993; Shires and others 2001). Many TCRαβ+ IELs express CD8αα together with CD4 or CD8αβ. CD8αα is composed of 2 CD8α subunits, and its expression level is proportional to its signal strength (Cheroutre and Lambolez 2008). However, it is not a TCR coreceptor; rather, CD8αα downregulates the functional avidity of the MHC-TCR activation complex (Cheroutre and others 2011). Type-a IELs are specific for non-self-antigen because of the positive and negative selection in the thymus. They can be detected in the blood, lymph, and secondary lymphoid organs, including the Peyer's patches.
Type-b IELS
Type-b IELs do not express either CD4 or CD8αβ; instead, the T cells are either CD8αα+ or CD8αα−, and they express TCRγδ or TCRαβ. First, the triple-positive thymocytes, which express CD4, CD8αβ, and CD8αα, differentiate into the double-negative TCRαβ+ cells that are the precursors of natural TCRαβ+ IELs. They then partially acquire their antigen-experienced phenotype during selection with self-antigens. However, they do not express CD2, CD5, CD28, or Thy1. An additional unique feature of IELs is that they can express NK receptors, and these IELs are referred to as natural killer T IELs (Guy-Grand and others 1996). The main T-cell populations in the IELs are TCRγδ+ T cells, which are believed to be fast acting, because they can be activated without professional antigen presenting cells. However, under normal conditions, TCRγδ+ T cells are tolerant against commensal bacteria. A recent study found that most of the TCRγδ+ T cells in the IEL are constitutively activated, as indicated by the expression of the activation marker CD69, whereas TCRγδ+ T cells in the lamina propria are negative for CD69 (Park and others 2010).
Cytokine Production by IELs
IELs have been proposed to play an important role in the regulation of intestinal epithelia by producing cytokines, which are key modulators of the intestinal mucosal barrier. They modulate important biological cellular functions that trigger downstream signaling pathways, and they mediate barrier homeostasis and injury. When the balance of Th1 [interferon-γ (IFN-γ), tumor necrosis factor (TNF), and interleukin (IL)-2] and Th2 (IL-4, IL-6, and IL-10) cytokines is broken, the intestinal barrier function is affected (Table 1). Recently, many studies have focused on the Th17 pathway mediated by IL-23 and IL-17, both of which play a key role in the inflammatory response (Yen and others 2006). Herein, we comprehensively analyze the broad spectrum of cytokines that are produced by IELs, including IFN-γ, TNF, IL-2, IL-4, IL-6, IL-10, transforming growth factor (TGF)-β, keratinocyte growth factor (KGF), and IL-17. Other cytokines, such as IL-1, IL-3, IL-5, and IL-13, can be secreted by IELs, but their role in the intestinal barrier function is rarely mentioned.

IFN, interferon; TNF, tumor necrosis factor; IL, interleukin; KGF, keratinocyte growth factor; TGF, transforming growth factor; Th1, T helper 1; IEL, intraepithelial lymphocyte; IEC, intestinal epithelial cell; MLC, myosin light chain; PI3K, phosphatidylinositol 3-kinase; TJ, tight junction; LPS, lipopolysaccharide; AJ, adherens junction; TFF, trefoil factor family; I/R, ischemia/reperfusion.
Interferon-γ
IFN-γ, or type II IFN, is a critical cytokine for innate and adaptive immunity against viral and intracellular bacterial infections and for tumor control. In the intestinal epithelia, the predominant source of IFN-γ is IELs. In the absence of in vitro stimulation, IELs produced low levels of CCL5, a prominent proinflammatory molecule in the intestine. Notably, higher levels of IFN-γ were produced by CD3 stimulation in the presence of CD43 costimulation than by CD3 stimulation alone (Montufar-Solis and others 2005). Systemic and local factors also regulate IEL-derived IFN-γ expression. Norepinephrine treatment significantly suppressed IFN-γ and TNF-α production by IELs via adrenoceptor β1, and histamine significantly decreased the production of Th1-cytokines (IFN-γ, TNF-α, and IL-2) and IL-4 in IELs (Takagaki and others 2009; Takayanagi and others 2012). Many adverse environmental factors directly or indirectly affect the function of IELs by regulating their expression of IFN-γ. The total parenteral nutrition (TPN)-induced IFN-γ expression resulted in an increase in Fas-L expression and EC sensitivity to Fas, with a resultant increase in EC apoptosis (Yang and others 2003). Repeated stress induced the overexpression of colonic IFN-γ, leading to larger amounts of phosphorylated myosin light chain (MLC), which altered the colonic mucosal barrier functions associated with bacterial translocation (Ferrier and others 2003). Further, McKay and others (2007) showed that IFN-γ-induced increases in epithelial paracellular and transcellular permeability were critically dependent on phosphatidylinositol 3-kinase (PI3K) activity. Recently, several studies have assessed the direct effect of IFN-γ on the expression and distribution of TJ proteins. IFN-γ stimulation decreased the levels of ZO-1 and perturbed the apical actin organization in T84 cells, leading to disruption of the TJs and an increase in paracellular permeability (Youakim and Ahdieh 1999). Moreover, exposure to IFN-γ resulted in the dramatic reorganization of TJ proteins at the epithelial lateral membrane, with internalization of the transmembrane proteins occludin, claudin-1, claudin-4, and junctional adhesion molecule-1. Contrarily, the authors observed that ZO-1 localization and expression were only slightly affected by IFN-γ-treatment. They hypothesized that ZO-1 maintains its localization to provide a scaffold for the efficient reassembly of TJ proteins upon cytokine withdrawal, which would be required for a rapid and critical reestablishment of epithelial barrier function (Bruewer and others 2003). Bruewer and others used selective inhibitors and markers to study the roles of 3 major internalization pathways in the disruption of TJs in IFN-γ-treated IECs. Inhibitors of macropinocytosis blocked the internalization of TJ proteins and junctional proteins that colocalized with macropinocytosis markers, dextran and phosphatidylinositol-3,4,5-trisphosphate. They also found that the internalized TJ proteins were found in early and recycling endosomes, but not in late endosomes/lysosomes (Bruewer and others 2005). More recently, a study showed that IFN-γ regulated E-cadherin stability by a Fyn kinase-dependent mechanism, which suggested a novel mechanism of EC contact disruption (Smyth and others 2012). Another study reported that lipid rafts were disrupted in IFN-γ-treated Caco-2 IECs, but this was not associated with a clear relocalization pattern of occludin or ZO-1 from the raft to nonraft fractions (Bowie and others 2012). More specifically, IFN-γ secreted by activated TCRαβ+ CD8αβ+, type-a, IELs disrupted the integrity of monolayers of the murine intestinal crypt-derived cell-line mIC (cl2) (Zufferey and others 2009). Nondestructively, IFN-γ induced LPS responsiveness by augmenting LPS uptake, MD-2 mRNA expression, and intracellular TLR4 protein levels (Suzuki and others 2003). IELs eliminated infected and senescent ECs through a combination of cytotoxicity and IFN-γ and TNF release (Guy-Grand and others 1998). In short, IFN-γ impacts the mucosal barrier through various molecules and cells and at different stages of the immune response. Importantly, it is the primary effector cytokine responsible for destroying the barrier.
Tumor necrosis factor
TNF is a monocyte-derived cytotoxin that has been implicated in tumor regression, septic shock, and cachexia. The first 2 members of the family to be identified were TNF, also known as TNF-α, and lymphotoxin-α, also known as TNF-β. The release of TNF-α is initially under strict feedback control by the uptake of free cytokines by IELs, and this is followed by a subsequent decline in transcription to avoid the accumulation of this potentially toxic cytokine (Ebert and Mehta 2006). Further, superantigens, including staphylococcal antigens (staphylococcal enterotoxin B, staphylococcal enterotoxin E, and toxic shock syndrome toxin 1), are potent inducers of IELs and induce TNF production (Sperber and others 1995). TNF-α is a central mediator of intestinal inflammation and is increased during human inflammatory bowel disease (IBD). In this model, early-onset colitis was dependent on TNF-α-preceded depletion of adherent and goblet cell mucin before EC damage (Dharmani and others 2011). Moreover, it was shown that enterocyte apoptosis was diminished in mice lacking TNF-α receptors and Fas, but there was no significant reduction in tissue damage (Merger and others 2002). This was unexpected, given that in vivo administration of TNF-α induced an enteropathy characterized by villus shortening (Guy-Grand and others 1998). In vitro, TNF-α is toxic to ECs, and both a TNF-R1 agonist and recombinant human TNF-α were cytotoxic when combined with IFN-γ, suggesting that the cytotoxicity is mediated through TNF-R1 (Sveinbjornsson and others 1997). Studies in cultured monolayers and animal models have demonstrated that TNF causes TJ barrier dysfunction via a process that requires MLC kinase (MLCK) activation. This is dependent on nuclear factor kappa B (NF-κB) because the inhibition of NF-κB or the silencing of NF-κB p65 prevented the TNF-induced increase of MLCK promoter activity in Caco-2 cells (Ye and others 2006; Weber and others 2010). Further, TNF activated MLCK to phosphorylate MLC in IECs and caused actomyosin contraction. These events led to TJ reorganization, caveolin-1-dependent occludin internalization, and increased TJ permeability (Shen 2012). Chemokines, particularly IL-8, are produced by ECs, and they likely recruit IELs and polymorphonuclear neutrophils into the epithelium. CD58 expression by HT-29 cells leads to increased TNF-α production via the CD2 pathway. TNF-α release, in turn, augments IL-8 synthesis and CD58 expression by the HT-29 cells (Ebert and others 2009). TNF-α was significantly increased in SBS (short bowel syndrome) mice, while it was decreased during ACE-I (angiotensin-converting enzyme inhibition) in a colitis model (Wildhaber and others 2005a). During an infection, IEL-derived TNF plays an important role in the mucosal barrier. The addition of IFN-γ, TNF-α, or supernatants obtained from cultured IELs from Eimeria vermiformis-infected mice reduced the transepithelial electrical resistance (TER) in a confluent CMT93 cell monolayer (Inagaki-Ohara and others 2006). TNF-α release by γδ+ IELs contributed to the upregulation of ferritin expression and possibly to the normal maintenance of the IEC apoptotic pathway (Ten Elshof and others 1999). Ebert (1998) noticed that TNF-α mainly affected intestinal CD8+ rather than CD4+ T cells and IELs rather than lamina propria lymphocytes (LPLs), and it increased the proliferation and migration of IELs, which may have led to their expansion in the epithelium. Conversely, Edelblum and others found that TNF administration reduced γδ+ IEL migration within the epithelial monolayers. This is likely because TNF leads to epithelial occludin endocytosis, and occludin is concentrated at the sites of the γδ+ IEL/epithelial interactions, where it forms a ring surrounding the γδ+ IELs (Edelblum and others 2012).
Interleukin-2
IL-2 is normally produced by T cells during an immune response. In rats, the CD4+CD8αα+ i-IELs produce IFN-γ and IL-2, while the CD4-CD8αα+ i-IELs produce abundant levels of TGF-β, but not IL-2, IFN-γ, or IL-4 (Yamada and others 1999). Age has a significant effect on the mucosal immune response of humans. The peripheral blood mononuclear cells and LPLs of older subject produce significantly less IL-2 than younger subjects. However, the ability of IELs to proliferate and produce IL-2 is not affected by age (Beharka and others 2001). IL-2 is necessary for the growth, proliferation, and differentiation of T cells to become effector T cells. IELs are often affected by several factors in the local environment. When IELs are exposed to stem cell factor combined with IL-2, there is an enhancement in the phosphorylation of JAK-3, and these 2 factors act synergistically to induce IEL proliferation, IFN-γ production, non-MHC-restricted cytotoxic activity, and the upregulation of gammac (the common γ-chain) (Wang and others 2000). There were significantly more IL-2-producing clones and fewer IL-10-producing cells in celiac disease samples compared to healthy samples. IL-2 can act on other cell types by increasing cytotoxic activity, cytokine production, Fas/FasL expression, and enterocyte apoptosis (Kolkowski and others 2006). In IL-2+/− mice, reduced levels of IL-2 led to attenuated activation and function of intestinal T cells and a failure to react adequately to DSS exposure (Sund and others 2005). During ischemia/reperfusion (I/R), the administration of IL-2 caused an influx of CD4+CD8+ immature IELs into the intestinal mucosa, which restored the IEL subset distribution and the mucosal barrier function (Nussler and others 2000). Therefore, we suggest that the influence of IL-2 on the intestinal mucosal barrier occurs predominantly through its effect on lymphocytes.
Interleukin-4
IL-4 is a cytokine that induces the differentiation of naïve helper T cells (Th0 cells) to Th2 cells. It has many biological roles, including the stimulation of B- and T-cell proliferation, and it also decreases the production of Th1 cells, macrophages, IFN-γ, and dendritic cell-derived IL-12. Although IELs produced significantly higher levels of IL-4 and Th1 cytokines (IFN-γ, TNF-α, and IL-2) when activated by PMA and ionomycin, activation by LPS did not have an effect on the IEL-derived cytokines tested (Takagaki and others 2009). Sepsis resulted in lower IEL γδ+ T-cell percentages and higher mRNA expression of IL-4, IFN-γ, TNF-α, IL-13, IL-17, retinoid acid receptor-related orphan receptor γt, and complement 5a receptor. Treatment with glutamine prevented the apoptosis of γδ+ IELs and downregulated their expression of inflammatory mediators, including IL-4 (Lee and others 2012). Intestinal flow diversion decreased the secretion of IL-4 and IFN-γ by IELs and LPLs. These results suggest an anti-inflammatory role of split ileostomy in patients suffering from IBD (Schmit and others 2000). As for the intestine, several studies have identified an association of high IL-4 tissue levels with an onset or relapse of inflammation. IL-4 has also been shown to act directly on the epithelial barrier function by decreasing TER and enhancing the endosomal area and paracellular spaces containing horseradish peroxidase, which depended on new protein synthesis and PI3K (Di Leo and others 2002). The incubation of T84 cells with IL-4 led to increased claudin-2 expression with corresponding decreases in transepithelial resistance and increases in permeability (Wisner and others 2008). Apart from the influence of intercellular junctions, IL-4-treated LS174T cells were able to produce mucins with a higher degree of O-glycosylation than untreated counterparts (Kanoh and others 2008). Using sodium azide (NaN[3]), an ATP synthesis inhibitor, Mochizuki and others (2009) found that fairly large molecules, even 150-kDa FITC-dextran, are able to permeate the intestinal epithelial monolayers via the energy-independent paracellular pathway upon exposure to excessive IL-4 (Mochizuki and others 2009). Th2 cytokines have been shown to downregulate the inflammatory response. Lugering and others found that the Th2 cytokines IL-4 and IL-13, but not IL-10, strongly downregulated IL-8 secretion from IECs, which partially altered the recruitment of immune cells to enterocytes. This inhibition of IL-8 might diminish the severity of the intestinal inflammatory response and thus reduce the clinical disease (Lugering and others 1999).
Interleukin-6
IL-6 is a pleiotropic cytokine with a central role in immune regulation, inflammation, hematopoiesis, and oncogenesis. As mentioned above, claudins 2 and 16 counteract epithelial resistance by creating cation-selective pores. Studies have found that IL-6 markedly induces claudin-2 expression in an MEK/ERK and PI3K-dependent manner (Suzuki and others 2011). IL-6 is a multifunctional NF-κB-regulated cytokine that acts on epithelial and immune cells and protects IECs from apoptosis, and this is largely mediated by the transcription factor Stat3. Therefore, the NF-κB-IL-6-Stat3 axis plays an important role in the proliferation and survival of tumor-initiating IECs (Grivennikov and others 2009). Further, IL-6 has been shown to rescue enterocytes from hypoxia-induced apoptosis, and IEC-6 cells that are resistant to apoptosis showed reduced Fas expression and increased Bcl-2 expression after coculture with LPS and IL-6 (Rollwagen and others 2006). In I/R injury, IL-6 is effective at protecting the intestine by maintaining graft blood flow and by reducing neutrophil infiltration and intragraft and circulating IL-1-β, TNF-α, and IL-6 (Kimizuka and others 2004). Contrarily, Wang and others showed that IL-6 and its signaling pathway may play a major role in the pathogenesis of many diseases. In IBD, IL-6 has been shown to be required for the development of Th1 cell-mediated colitis. Basolateral IL-6 stimulation resulted in a maximal induction of NF-κB activation and polarized expression of ICAM-1, an adhesion molecule shown to be important in the neutrophil–epithelial interactions (Wang and others 2003). The roles of IL-6 in different disease models must be distinguished carefully.
Interleukin-10
IL-10 is often considered the most important anti-inflammatory cytokine in humans and is secreted by a variety of cells. Both αβ-TCR+ IEL and γδ-TCR+ IEL populations express IL-10, and this expression is significantly increased in the presence of IL-7 (Yang and others 2005). Another study showed that human IELs from the jejunal mucosa produced massive quantities of IL-10 and IFN-γ after 3 or 10 days of culture with IL-15, and that endogenous IL-10 promoted FL-mediated cytotoxicity (Ebert 2005). The vitamin D receptor (VDR) mediates T-cell homing to the gut. The VDR KO mouse had reduced numbers of CD8αα+ IELs, which coincided with low levels of IL-10 in the small intestine. This resulted in an increased inflammatory response to the normally harmless commensal flora (Yu and others 2008). The addition of Saccharomyces boulardii to IEL cultures stimulated by Escherichia coli and Candida albicans increased the anti-inflammatory IL-10 and IL-4 responses, because S. boulardii has been shown to be protective against diarrheal pathogens by reducing proinflammatory responses (Fidan and others 2009). Forsberg and others divided IELs into 3 major subsets: γδ+ IELs, CD4+αβ+ IELs, and CD8+αβ+ IELs. The production of IL-10 is a common feature of these IEL subsets in celiac disease, and they therefore attempt to limit inflammation in an autocrine fashion (Forsberg and others 2007). Conversely, it has been reported that human intestinal αβ+ IEL clones in celiac disease show reduced IL-10 synthesis, which is thought to be due to single-nucleotide polymorphisms of the IL-10 promoter, although there is currently insufficient evidence for this (Kolkowski and others 2006). During Cryptosporidium parvum infection, there is a temporal association between the expression of IL-10 by IELs and the expression of C. parvum antigens in the infected calf epithelium (Wyatt and others 2002). A recent study revealed that IL-10 knockout mice suffer from spontaneous colitis, and they exhibit increased paracellular permeability in conjunction with decreased expression and redistribution of zonula occludens-1, occludin, claudin-1, and β-catenin (Chen and others 2010). Additionally, a study from our group found that IEL-derived IL-10 could contribute to TPN-associated barrier loss, and exogenous IL-10 administration significantly attenuated the TPN-associated decline in ZO-1, E-cadherin, and occludin expression, as well as the loss of intestinal barrier function (Sun and others 2008). Administration of tunicamycin to wild-type mice caused intestinal endoplasmic reticulum (ER) stress and accumulation of misfolded Muc2 of goblet cells. However, IL-10 could prevent protein misfolding and ER stress by maintaining mucin production and help the intestine preserve the mucus barrier (Hasnain and others 2012). Undesired immunosuppressive effects and the invasion of pathogens can be observed in diseases with IL-10 overproduction. In diseases with IL-10 deficiency, persistent immune activation occurs, and this in turn can cause local tissue damage.
Transforming growth factor-β
TGF-β exists in 3 known subtypes in humans: TGF-β1, TGF-β2, and TGF-β3. It is well known that TGF-β1 is secreted by multiple T cells, and CD4-CD8αα+ type-b IELs, but not CD4+CD8αα+ IELs, produce abundant levels of TGF-β (Yamada and others 1999). Denning and others found that TCRαβ+ CD8αα+ IELs constitutively synthesized TGF-β1 mRNA, which was not augmented after Ag-specific stimulation. However, the stimulation did increase the expression of TGF-β3 mRNA (Denning and others 2007). Importantly, TGF-β3 enhanced intestinal epithelial healing in an in vitro model system (McKaig and others 1999). It has been shown that TGF-β1 affects claudin-4 gene expression via Smad-4-dependent and Smad-4-independent transcriptional regulation, resulting in barrier protection (Hering and others 2011). In the absence of enteral nutrition, TPN resulted in increased TGF-β1 expression by IELs, which was associated with villous atrophy and increased IEC apoptosis (Wildhaber and others 2005b). N-3 polyunsaturated fatty acid (PUFA) slightly increased the expression of TGF-β1 mRNA, but significantly decreased the expression of TNF-α, IFN-γ, IL-4, and IL-10 mRNA. This could be attributed to the immunosuppressive effect of N-3 PUFA (Wang and others 2008). As the β7-integrin gene (Itgb-7) promoter is responsive to TGF-β1, local TGF-β upregulated CD103 on CD8+ IELs and contributed to their adherence to epithelial E-cadherin. This demonstrated that TGF-β signaling plays a role in the retention of lymphocytes within the intestinal epithelium (Lim and others 1998; Baker and others 2006). TGF-β also inhibited the mitosis of IELs to mitogens, IL-7, and stimuli of the CD2 and CD3 pathways (Ebert 1999). Furthermore, TGF-β attenuated epithelial damage and TER changes that were induced by supernatants of IEL cultured from E. vermiformis-infected mice. IFN-γ and TNF-α were thought to be the main components of these supernatants (Inagaki-Ohara and others 2006). The transfer of γδ+ IELs suppressed the mortality rate and the severity of TNBS-induced colitis due to their production of TGF-β (Inagaki-Ohara and others 2004).
Keratinocyte growth factor
KGF, a member of the fibroblast growth factor family, is a known mitogenic growth factor and has been shown to stimulate the proliferation of a variety of EC lines. KGF is strongly expressed throughout the gastrointestinal tract, suggesting that γδ + IELs, which are the only source of KGF, function in surveillance and repair of damaged epithelial tissues. Using the villus atrophy TPN mouse model and the villus hypertrophy SBS mouse model, our group demonstrated that TPN administration was associated with a downregulation of IEL-derived KGF expression, and short bowel syndrome was associated with an upregulation of IEL-derived KGF expression. IEL-derived KGF expression was highest in the crypts, was slightly less in the lower portion of the villi, and was markedly lower in the upper portion of the villi. These results showed that KGF from IELs is an important factor for the maintenance of IEC proliferation and villus growth (Yang and others 2004). Further, KGF increased Bcl-2 expression and other antiapoptotic family members and decreased TPN-induced EC apoptosis (Wildhaber and others 2003). γδ-deficient mice (TCRδ−/− ) and KGF-deficient mice (KGF−/− ), but not αβ-deficient mice (TCRα−/− ), were more prone to DSS-induced mucosal injury than wild-type mice and demonstrated delayed tissue repair after termination of the DSS treatment (Chen and others 2002). IL-8 is a potent proinflammatory cytokine, yet it may also play a role in wound healing by inducing angiogenesis and IEC migration. KGF may also play a role in enhancing the IL-1-stimulated production of IL-8 by ECs during mucosal inflammation (Unger and McGee 2011). Further, it was shown that KGF markedly increased goblet cell numbers and TFF3 expression throughout the intestine, despite a short-term lack of food intake. In addition, KGF induced ectopic TFF2 expression in proximal small-bowel goblet cells (Fernandez-Estivariz and others 2003). Tritiated thymidine or bromodeoxyuridine labeling showed statistically significant increases in labeling in the stem cell zone of the crypt by protracted exposure to KGF, with a concomitant reduction in labeling in the upper regions of the crypt corresponding to the late-dividing transit population. Another action of KGF is to increase intestinal crypt stem cell numbers or increase the number of stem cells in the S phase of the cell cycle (Potten and others 2001). Recently, our group found that I/R resulted in the disruption of TJ proteins (claudin-1 and ZO-1), and KGF could attenuate this damage (Cai and others 2012a). In addition, we found that crosstalk between IELs and ECs exists, and KGF can regulate EC-derived IL-7 expression. This regulation is likely associated with the protective effects of KGF on intestinal injury (Cai and others 2012b). Collectively, the γδ+ IEL-derived KGF is crucial for restoring the integrity of the epithelium in response to the physical and inflammatory damage.
Interleukin-17
Similar to IFN-γ, IL-17 acts as a potent mediator in delayed-type reactions by increasing chemokine production in various tissues to recruit monocytes and neutrophils to the site of inflammation. In addition to IL-17A, the members of the IL-17 family include IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F, and they all have similar protein structures. IL-17 expression is increased during colitis, and it is known that IL-17A is predominantly produced by the Vγ4 subset of TCRγδ+ T cells. Park and others (2010) found that the expansion of IL-17A-expressing TCRγδ+ IELs was responsible for colitis induction, presumably through the secretion of IL-17. In active CD cells, γδ+ IELs expressed IL-17A, but to a lesser extent than γδ− IELs (Monteleone and others 2010). The IL-17-induced formation of TJs correlated with the upregulation of the claudin-1 and claudin-2 gene transcription. Claudin-2, but not claudin-1, is regulated by IL-17 through the ERK/MAPK (extracellular signal-related, mitogen-activated protein kinases) pathway in T84 cells (Kinugasa and others 2000). Therefore, IL-17A could play a role in maintaining or expanding tissue-damaging inflammatory reactions. The most recent study reported that IL-17 and IL-17C treatment of YAMC cells enhanced mRNA expression of occludin, claudin-1, and claudin-4. Interestingly, IL-17 and IL-17C was shown to be protective against the development of DSS-induced colitis. Thus, future study should focus on the role of the IL-17 family in regulating intestinal inflammation, as well as on the mechanisms promoting the mucosal barrier integrity (Reynolds and others 2012).
Conclusions
The suggested functions of IELs are the mediation of inflammatory reactions, surveillance of the intestinal epithelium, and the induction or maintenance of oral tolerance. It is plausible that IELs also play a crucial role in the development and progression of intestinal diseases (Fig. 1). Specifically, cytokines are one of the major ways in which IELs affect the intestinal barrier function. Different cytokines can regulate the surrounding cells, EC antigen-presenting function and survival, and paracellular permeability, which involves TJs, adherens junctions, gap junctions, and DMs. The assembly and stability of these molecules are modulated by the intracellular signals transduced by cytokines. The local intestinal immune response is also subject to the effects of these cytokines. IFN-γ, TNF-α, and IL-2 produced by IELs have the potential to initiate or propagate gut inflammation or lead to an aberrant immune response. However, IELs express a large number of regulatory cytokines, such as IL-4, IL-10, and TGF-β. These cytokines display regulatory functions and prevent excessive inflammatory responses that could jeopardize the integrity of the intestinal barrier. Further studies are needed to distinguish the contribution made by IEL-derived cytokines in the inflamed intestine from that of systemic cytokines. Much work needs to be done to decipher the communication networks between IELs and the systemic immune system.
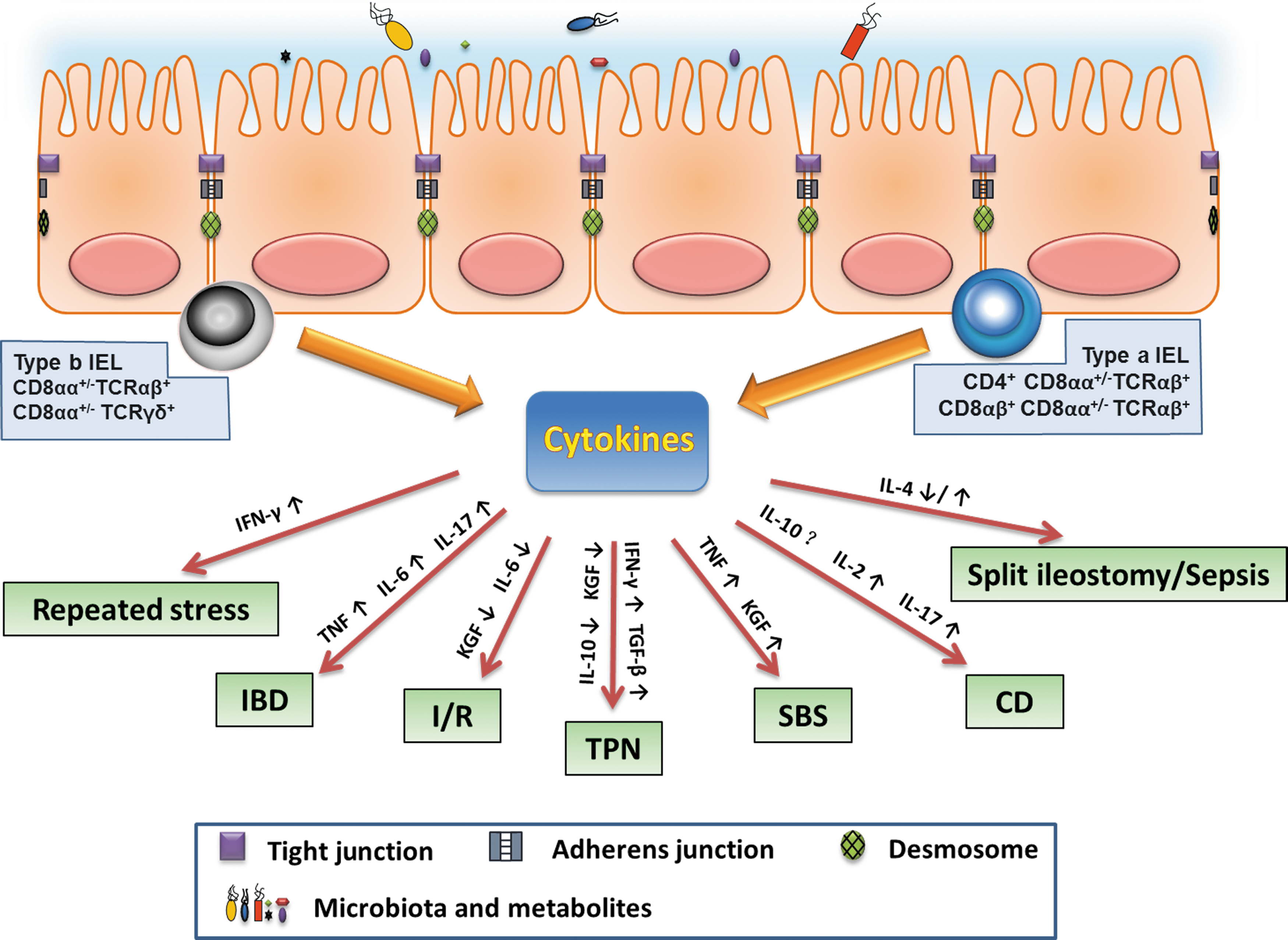
Schematic overview of the principal cytokines produced by intraepithelial lymphocytes (IELs) involved in different intestinal diseases. IEL-derived cytokines play an important role in mediating barrier homeostasis and injury. A certain disease, may involve multiple IELs' cytokines dysregulation. IBD, inflammatory bowel disease; I/R, ischemia/reperfusion; TPN, total parenteral nutrition; SBS, short-bowel syndrome; CD, celiac disease; IFN, Interferon; TNF, tumor necrosis factor; IL, interleukin; KGF, keratinocyte growth factor; TGF, transforming growth factor.
Footnotes
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No 30973113, 81020108023, and 81000830) and the Chongqing Science and Technology Commission International Key Collaboration Project (CSTC 201110008).
Author Disclosure Statement
No competing financial interests exist.