
Abstract
Cardiovascular diseases are a major cause of morbidity and mortality worldwide. The interferon inducible transcriptional activator signal transducer and activator of transcription-1 (STAT1) and p53 are two critical transcriptional factors that have pivotal roles in cardiac biology and pathology. Here we describe a novel interplay between these two key players that critically regulate the levels of the pleiotropic interleukin 6 (IL6) in the heart. We provide in vivo evidence to demonstrate that, in cardiac tissues, STAT1 is a positive regulator of IL6 expression and it competes with the suppressive effect of p53 to sustain basal IL6 levels. Induction of IL6 expression in response to interferon gamma (IFNγ), a well-characterized activator of STAT1, parallels that of STAT1 phosphorylation and induction of STAT1 target genes, such as the interferon regulatory factor-1 (IRF-1), major histocompatibility complex class II transactivator (C2ta), and β2-microglobulin (B2m). Furthermore, hearts from STAT1 knockout mice fail to induce IL6 expression in response to IFNγ. More importantly, we showed that this regulatory system is not functional in mouse embryonic fibroblasts, suggesting that activation of IL6 expression by STAT1 may be tissue specific. IL6 is a major effector of inflammation and cardiac hypertrophy, two major processes involved in heart failure, and therefore, understanding the molecular mechanisms regulating IL6 expression will enable better therapies and treatments for cardiovascular disease patients.
Introduction
Interleukin 6 (IL6) is a pleiotropic cytokine that is highly expressed in cardiac myocytes and its expression is induced following I/R injury (Kukielka and others 1995). IL6 protein expression has been shown to increase in cardiac myocytes following hypoxic stress and this is augmented by reperfusion (Yamauchi-Takihara and others 1995). The IL6/soluble IL6 receptor complex reduces the infarct size in rats following I/R injury (Matsushita and others 2005). Induction of IL6 by hypoxia in NRCMs has been demonstrated to be mediated by the transcription factor, nuclear factor-κB (NF-κB) (Matsui and others 1999). In human monocytic cells, induction of the IL6 gene by interferon gamma (IFNγ) and TNFα involves cooperation between the interferon regulatory factor-1 (IRF-1) and NF-κB (Sanceau and others 1995). Although STAT1 has not been directly implicated, it is a primary mediator of the IFNγ signaling pathway and IRF-1 is positively regulated by STAT1.
Here we showed that STAT1 is a critical regulator of basal IL6 expression in the heart and it can critically block the suppressive effect of p53 on cardiac IL6 expression. Furthermore, we demonstrated that IFNγ fails to induce IL6 expression on STAT1−/− background. Importantly, this regulatory system is not functional in MEFs, suggesting that the activation of IL6 expression by STAT1 is potentially heart specific.
Materials and Methods
Mouse models
All protocols involving animals were performed in accordance with the United Kingdom, Home Office Animals (Scientific Procedures) Act 1986.
p53 (B6.129-Trp53tmBrd N12) and STAT1 (129S6/SvEvTac-STATtm1Rds ) knockout mice were purchased from Taconic-USA. Eight-week-old mice were injected intraperitoneally with 10–200 ng recombinant mouse IFNγ (Sigma; I4777). MEFs were isolated from E13.5 embryos and cultured in DMEM containing 10% FBS.
Proteomic profiling of mouse hearts
Whole mouse hearts were snap-frozen in liquid nitrogen and homogenized in a lysis buffer (8 M Urea, 0.2% DTT, 0.5% CHAPS, and protease inhibitors). The 2D Quant Kit (Amersham) was used to determine the protein concentration according to the manufacturer's instructions. Immobiline DryStrip gel strips were used to separate proteins based on their isoelectric point using an Ettan IPGphor II isoelectric focusing (IEF) system (Amersham). Following IEF, proteins were resolubilized and carboamidomethylated, denatured in the urea buffer (6 M Urea, 50 mM Tris-HCl pH 8.8, 30% glycerol, 2% SDS, and 1% DTT), equilibrated in the Tris buffer (50 mM Tris-HCl pH 8.8, 30% glycerol, 2% SD,S and 2.5% iodoacetamide), and separated by SDS-PAGE. SDS-PAGE gels were fixed in 50% methanol, 10% acetic acid solution, and stained with silver nitrate. Gels were scanned using a GS-800 Densitometer (Bio-Rad).
Quantitative analysis
Cardiac proteomes were analyzed by using ImageMaster™ 2D Platinum software (Amersham). The selection criteria were set to a minimum area of 2, smoothness of 5, and saliency of 150. The differences in cardiac proteomes of wild-type (WT), p53−/− , STAT1−/− , and p53−/−/STAT1−/− mice were quantified by comparing the ratio of percentage volume densities between identified spots. ImageMaster selected proteins that had an average ratio >2 and did not have overlapping class intervals as significant changes and regarded proteins with the largest gap between class intervals as the most significant. Manual analysis of 2D gels was also used to verify proteins that were identified by ImageMaster as authentic protein spots.
Peptide mass fingerprinting
Protein spots were excised from gels and the gel pieces were dehydrated by incubation with acetonitrile for 20 min followed by centrifugal evaporation at 37°C. Access iodoacetamide was removed by repeated washes with 100 mM ammonium bicarbonate. The dehydrated gel pieces were incubated with 12.5 ng/mL trypsin in 50 mM ammonium bicarbonate overnight for in-gel trypsin digestion. Peptides were released from the gel matrix by using 50% acetonitrile containing 0.1% TFA and were transferred to fresh siliconized microfuge tubes. Hydrophobic peptides were extracted by using 4 M urea and were combined with acetonitrile extracted peptides before desalting. Zip-Tip C-18 microcolumns (Millipore) were used for peptide desalting according to the manufacturer's instructions.
Mass spectrometry
The α-cyano-4-hydroxycinnamic acid (#70990; Fluka) was dissolved in the acetonitrile: ethanol solution and mixed with 50 mM Fucose (Biochemika) in a 1:1 ratio. Peptide samples, α-cyano-4-hydroxycinnamic acid, and ACTH & Angiotensin II standards were mixed in a ratio of 3:1:1 and were spotted on MALDI-TOF plates. Samples were analyzed on a MALDI TOF MS (TOF Spec E; MicroMass). Peptide analyses were performed in a positive ion mode and spectra were acquired by averaging over a period of 5 scans of the highest signal. Data were acquired in a reflectron mode, operating over a mass range of 6,000 m/z with matrix suppression set out at 650 Da, and analyzed using Mass Lynx software (MicroMass).
Antibodies and immunoblot analysis
Snap-frozen whole mouse hearts were homogenized in the RIPA buffer (0.75 M NaCl, 5%NP40, 2.5%deoxycholate, 0.5%SDS, 0.25 M Tris-HCl pH 8.0, 10 mM DTT, and protease inhibitors). Proteins were denatured, separated on SDS polyacrylamide gels, and then transferred to nitrocellulose membranes. Following protein transfer, blots were incubated in the blocking solution and then with a primary antibody for 1 h. Antibodies were used at dilutions 1:1,000 STAT1Y701 (Zymed/Invitrogen) and 1:500 β-ACTIN (Santa Cruz). Immunoreactive proteins were detected using enhanced chemiluminescence (Amersham).
Quantitative real-time PCR
RNA extraction and cDNA synthesis were done using standard methods. The following real-time PCR primers were used: IL6 sense 5′ GAACAACGA TGATGCACTTGCAG 3′; IL6 antisense 5′ CCTTAGCCAC TCCT TCTGTGAC 3′; β-Actin sense 5′ AGATCACCCAGA TCATGTTTGAG 3′; β-Actin antisense 5′ AGGTCCAGAC GCAGGATG 3′; β2-micro sense 5′ TGCTGTCTCC ATGTT TGATGTATCT; β2-micro antisense 5′ TCTCTGCTCCCC ACCTCTAAGT 3′; GAPDH sense 5′ AATGTGTCCGTCGTG GATCTGA 3′; and GAPDH antisense 5′ GATGCCTGCTTC AC CACCTTCT 3′.
Enzyme-linked immunosorbent assay
The READY-SET-GO mouse IL6 kit (88-7064-88; Bioscience) was used for the enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions. Absorbance was measured at 450 nm using a Genios microplate reader (Tecan).
Statistical analyses
Statistical tests were performed using Graphpad Prism 4.0 software.
Results
Absence of p53 induces IL6 expression in the heart
As both p53 and STAT1 are critical for the normal physiology and pathology of the heart, we wanted to characterize the signaling pathways that are regulated by these key transcription factors by a proteomic approach.
To analyze the consequences of the loss of both p53 and STAT1 to cardiac proteome, STAT1−/− and p53−/− mice were crossed to generate p53−/−/STAT1−/− mice. The double knockout (KO) mice were viable and fertile, with a normal Mendelian ratio of genotypes in the offspring. However, these mice were susceptible to the spontaneous development of a variety of tumors and either died or had to be culled, because of sick health, at less than 3 months of age. Western blotting confirmed that these knockout mice lacked p53 expression and they expressed a C-terminal truncated form of STAT1 in cardiac tissues.
Cardiac protein samples from 8–10-week-old WT, p53−/−, STAT1−/− , and p53−/−/STAT1−/− mice were separated using IPG acrylamide strips of narrow-range pH 2D-PAGE (pH 4–7) and analyzed using ImageMaster software. Among the proteins that showed differential expression between the experimental groups, abundance of one protein was significantly upregulated in p53−/− hearts compared to all other groups (Fig. 1A, B). This protein had an isoelectric point (pI) of pI 7.0 and a mass of 20–25 kDa. By using peptide mass fingerprinting and MS-FIT database search, we identified this protein to be the IL6 precursor. The IL6 precursor has a pI of 7 and a molecular weight of 24384 daltons, thus matching with the predicted pI and molecular mass based on the separation from the 2D gels (Fig. 1B).
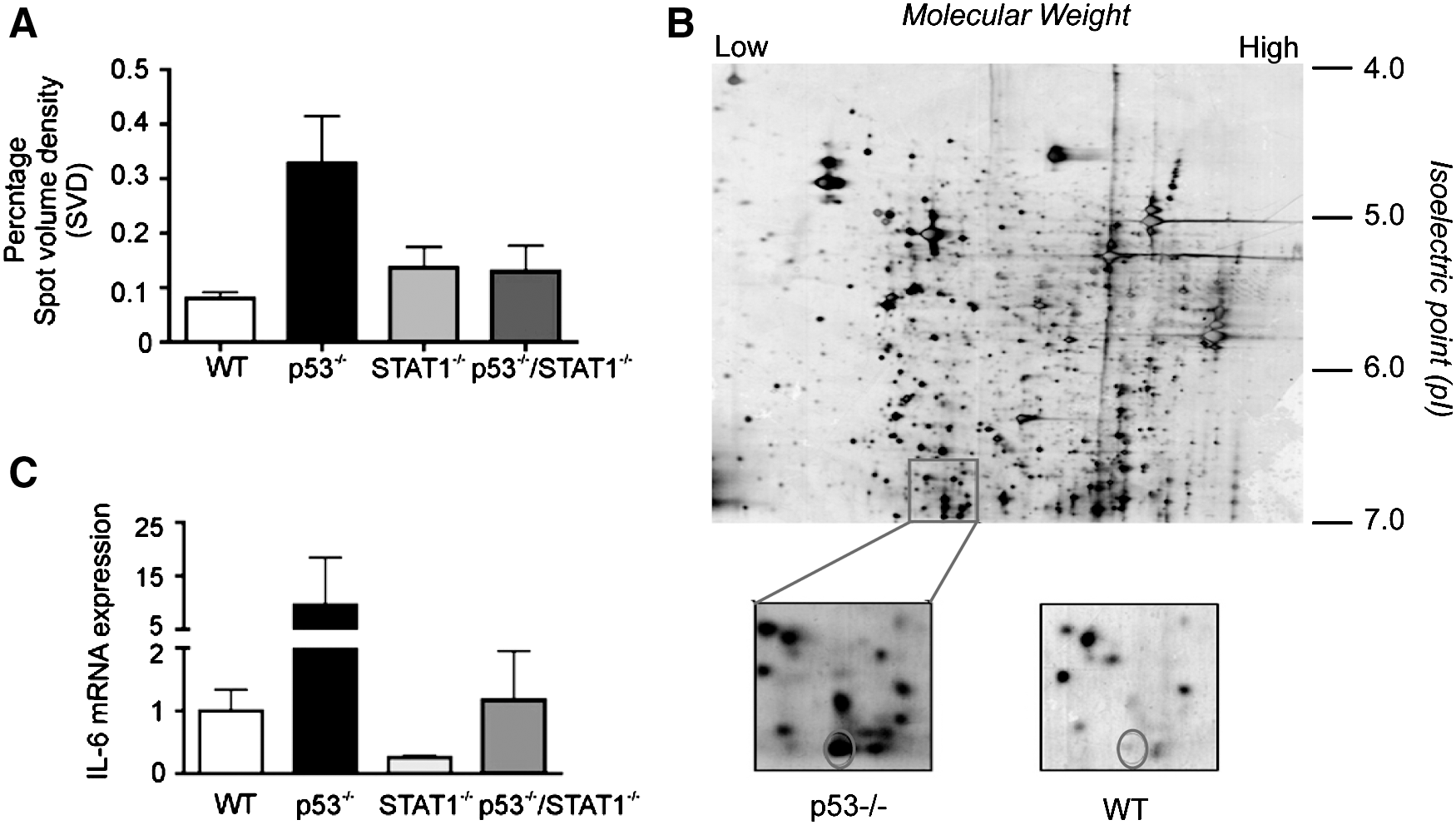
Interleukin 6 (IL6) is highly abundant in p53−/−
hearts.
To verify the role of p53 and STAT1 in regulating IL6 precursor expression in cardiac tissue, we analyzed IL6 mRNA levels in the hearts of p53−/−, STAT1−/− , and p53−/−/STAT1−/− mice, by quantitative real-time PCR (qRT-PCR). As shown in Fig. 1C, the loss of p53 expression resulted in a significant increase in IL6 precursor transcript levels, compared to the WT mouse heart. These data also support the previous findings on the suppressive role of p53 on IL6 expression (Santhanam and others 1991; Angelo and others 2002). Interestingly, we also observed an ∼4-fold reduction in IL6 mRNA levels in STAT1−/− hearts, suggesting that STAT1 may potentially be important for IL6 expression in cardiac tissues (Fig. 1C). Furthermore, IL6 mRNA levels in p53−/−/STAT1−/− mice were comparable to that of WT mice. These data suggest that the effect of STAT1 in promoting IL6 expression may potentially dominate over p53's suppressive effect and substantiates STAT1 as a critical regulator of basal IL6 expression in the heart.
STAT1 induces IL6 expression in the heart
To further investigate the role of STAT1 in the regulation of IL6 expression in the heart, we sought to induce the STAT1 activity by IFNγ and compare IL6 levels in WT and STAT−/− mice. The optimal dose and exposure time of IFNγ, which is sufficient to induce STAT1 activation, was determined by the time course and dose–response assays in WT mice. Activation of STAT1 was assessed on the basis of its phosphorylation at tyrosine 701, which is a prerequisite for its dimerization and nuclear translocation.
For the dose-curve assays, 8-week-old WT mice were injected intraperitoneally with 1, 10, 50, 100, or 200 ng/mL IFNγ or vehicle control, and cardiac proteins were extracted 2 h after injection. As shown in Fig. 2A, STAT1Y701 phosphorylation increased in a dose-dependent manner and maximum phosphorylation was observed when 200 ng/mL IFNγ was used. Based on this finding, the time curve for STAT1 activation was performed by using 200 ng/mL IFNγ and cardiac lysates were prepared 1, 2, 4, or 24 h after injection. While STAT1Y701 phosphorylation was evident for up to 2 h following IFNγ treatment, it was rapidly lost by 4 h (Fig. 2B).
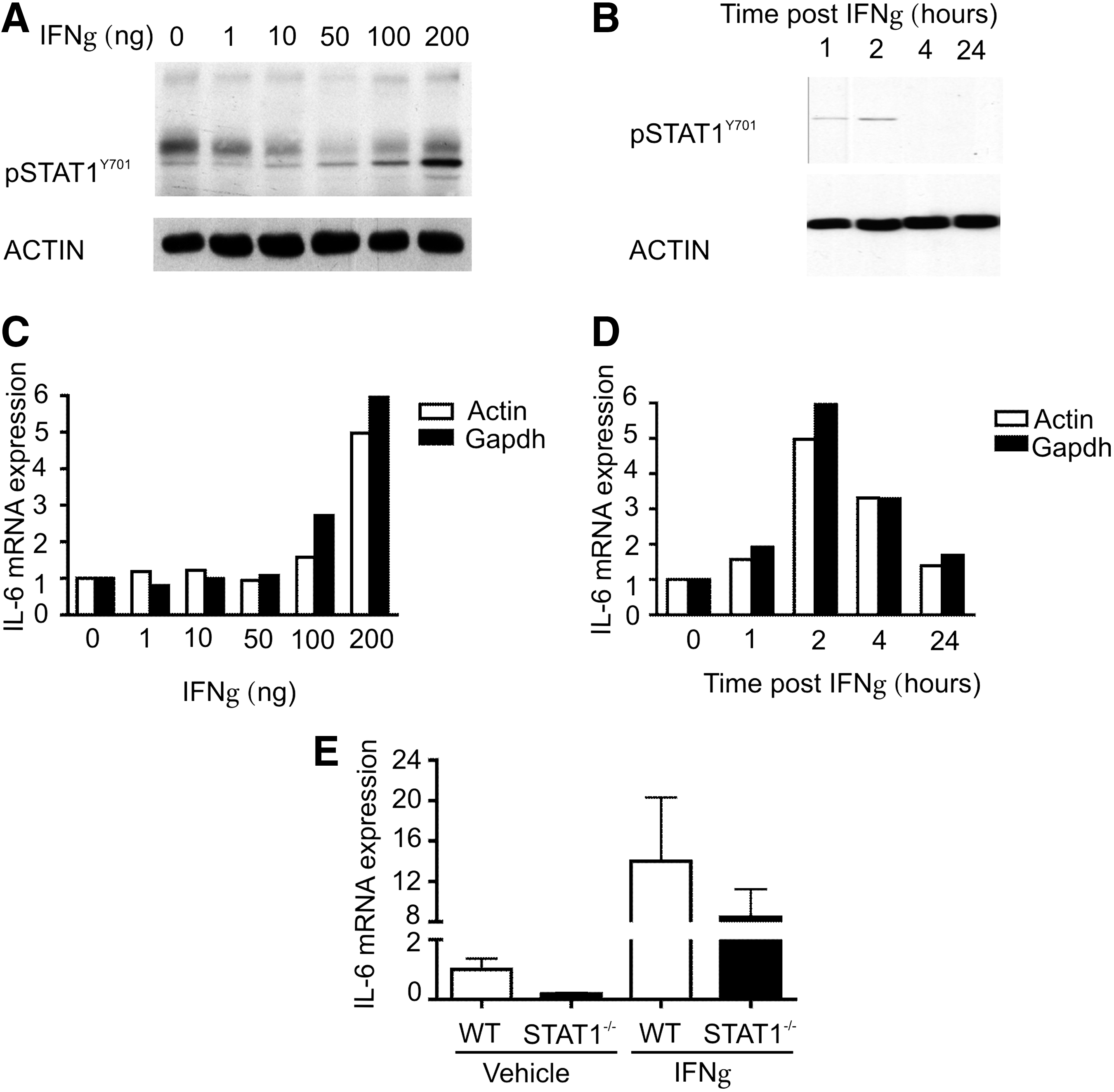
Signal transducer and activator of transcription-1 (STAT1) drives IL6 expression in the heart.
Activation of STAT1 was also assessed indirectly, by analyzing the transcriptional upregulation of well-characterized STAT1 target genes, including IRF-1, major histocompatibility complex class II transactivator (C2ta), and β2-microglobulin (B2m). In parallel with STAT1 phosphorylation, expression of IRF-1, C2ta, and B2m increased following IFNγ in a dose-dependent manner and maximal induction was observed with 200 ng/mL of IFNγ (Supplementary Fig. S1; Supplementary Data are available online at
Strikingly, kinetics of IL6 expression following IFNγ treatment paralleled that of STAT1 phosphorylation and transactivation of STAT1 target genes, both in the dose–response (Fig. 2C) and time-course (Fig. 2D) experiments. IL6 mRNA expression was highest in response to the maximum dose of IFNγ used and the peak IL6 expression was observed 2 h after IFNγ treatment (Fig. 2D). In accordance with these findings, induction of IL6 expression was considerably affected in STAT1−/− mice following IFNγ treatment (Fig. 2E), further substantiating IL6 as a candidate STAT1 target gene in the heart.
STAT1 fails to induce IL6 expression in MEFs
To validate our findings in another experimental system, we used MEFs isolated from p53−/− and p53−/−/STAT1−/− mice. MEFs from WT and STAT1−/− mice were excluded from experiments as they became senescent within 8 days in culture. Unexpectedly, the baseline IL6 transcript levels and secreted IL6 levels were higher in p53−/−/STAT1−/− MEFs compared to p53−/− MEFs (5-fold and 2-fold, respectively) (Fig. 3A, B).

STAT1 fails to induce IL6 expression in mouse embryonic fibroblasts (MEFs).
The loss of STAT1 expression in p53 −/− mice is associated with increased susceptibility to development of spontaneous tumors (compared to p53−/− mice) and enhanced expression of IL6 has been observed in different types of cancers (Paule and others 2000). These data suggest that the increase in IL6 expression and secretion we observed in STAT−/− MEFs could be due to the increased growth rate of fibroblasts. However, the MTS assays revealed that the growth rates of p53−/− and p53−/−/STAT1−/− MEFs were highly similar when grown in either low or high serum (data not shown). Therefore, the increased expression and secretion of IL6 in p53−/−/STAT1−/− mice, compared to p53−/− mice, were not due to the differences in growth rates of MEFs.
STAT1 has a repressive effect on IFN
γ
/IL6 signaling in MEFs
To provide further evidence to support our finding that STAT1 does not function upstream of IL6 in MEFs, we undertook an indirect approach by inducing the STAT1 activity in MEFs via IFNγ treatment.
Initially, we identified the optimal concentration and time for IFNγ treatment in p53−/− MEFs, as they contain STAT1 and are responsive to IFNγ. Tyrosine phosphorylation of STAT1 was assessed by Western blotting and the relative expression of β2m was determined by qRT-PCR. A dose of 5 ng/mL IFNγ was sufficient to induce STAT1Y701 phosphorylation within the first hour after treatment, however, maximum STAT1 activation was observed with 40 ng/mL IFNγ (Fig. 4A). As STAT1 phosphorylation was still significantly evident 4 h after IFNγ treatment, induction of β2-microglubulin was tested at this time point, both in p53−/− and in p53−/−/STAT−/− MEFs. As expected, β2-microglubulin transcript levels increased gradually along with increasing concentrations of IFNγ in p53−/− MEFs and no induction was observed in p53−/−/STAT−/− MEFs (Fig. 4B).
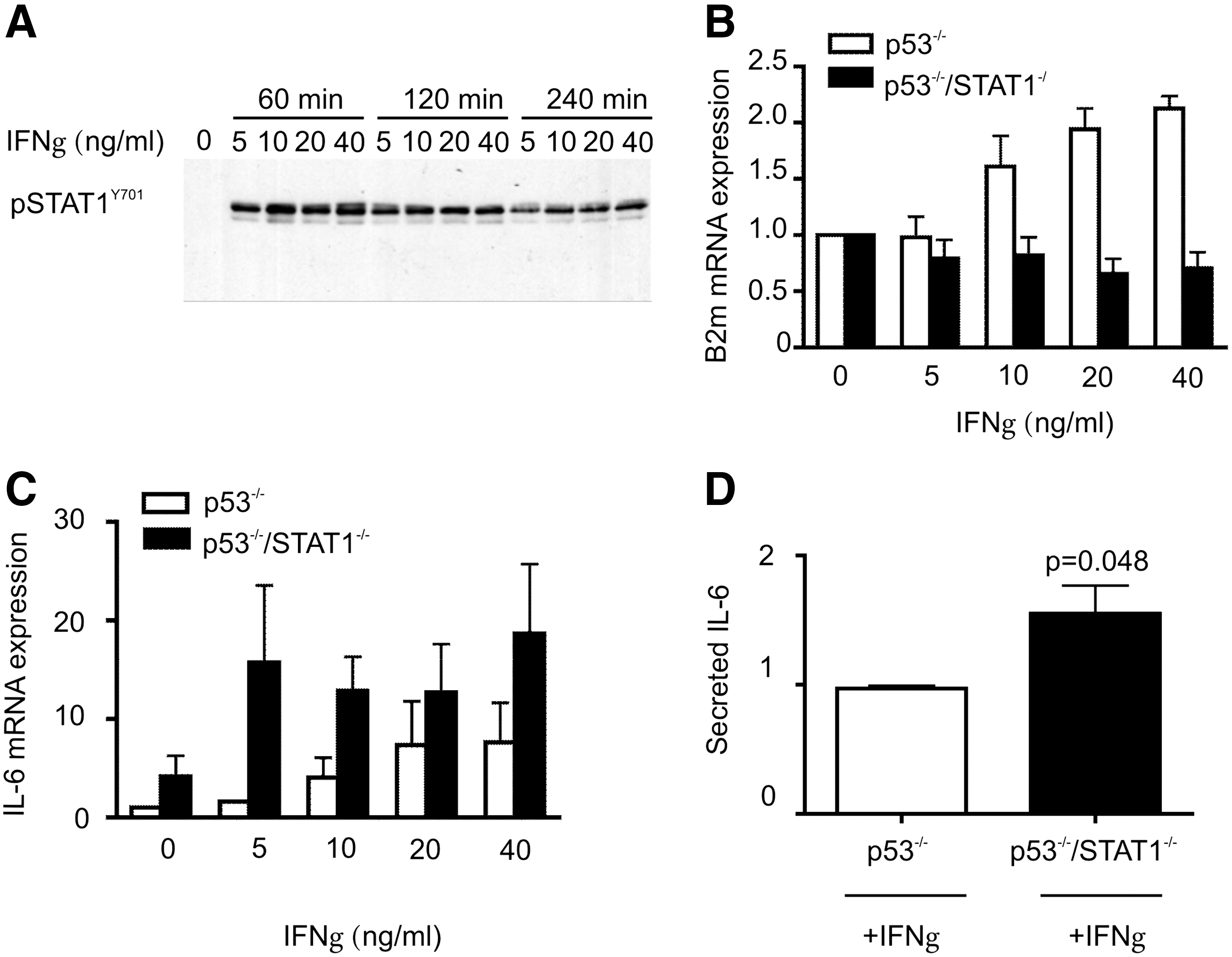
STAT1-deficient MEFs express more IL6 in response to IFNγ.
Under the same experimental conditions, IFNγ treatment resulted in up to 12-fold induction in IL6 expression in p53−/− MEFs (Fig. 4C). However, this effect was much more significant in p53−/−/STAT−/− MEFs, resulting in an ∼25-fold induction with 40 ng/mL IFNγ treatment. Furthermore, the amount of secreted IL6 in the culture media was also significantly higher in p53−/−/STAT−/− MEFs, compared to p53−/− MEFs, further providing evidence to support our findings with unstimulated MEFs (Fig. 4D).
These data demonstrate that STAT1 fails to induce IL6 expression in MEFs, and furthermore, also suggests that STAT1 may have a suppressive effect on IL6 expression in MEFs.
Discussion
The transcription factor STAT1 plays pivotal roles in cardiac biology. Although it induces expression of the majority of its target genes by directly binding to their promoters, recent evidence has demonstrated that it can also induce gene expression by acting as a transcriptional coactivator. In this regard, STAT1 has been shown to associate with the transcription factor p53 for maximal induction of p53 target genes. This interaction is particularly critical for induction of apoptosis in isolated neonatal cardiac myocytes following I/R-induced injury, and apoptosis is significantly inhibited in STAT1-deficient myocytes.
Based on this critical role of STAT1 and p53 in cardiac biology, we sought to determine the changes in cardiac proteome that are implemented by the absence of these transcription factors. For this purpose, we crossed STAT1 (129S6/SvEvTac-STATtm1Rds ) and p53 (B6.129-p53tmBrd N12) knockout mice to generate the STAT1/p53 double KO mice. The transgenic mice, with the disrupted STAT1 gene, express a truncated STAT1 protein of 72 kDa in the heart, a finding that has been previously described (Meraz and others 1996; Stephanou and others 2002). Although the p53−/−/STAT−/− mice were viable, the majority of them were susceptible to the spontaneous development of a variety of tumors at an early age. A previous study has also demonstrated that p53−/−/STAT1−/− mice develop a wide variety of tumors, with increased frequency at an earlier age compared to p53−/− mice (Kaplan and others 1998).Therefore, these mice either died or had to be culled because of ill health between 3 and 6 months of age.
Comparison of the cardiac proteomes of p53−/−, STAT1−/−, and p53−/−/STAT1−/− mice by 2D-PAGE analysis revealed that the IL6 precursor is differentially expressed among these genotypes in the heart. IL6 is a pleiotropic cytokine that is involved in antibody production, inflammation, and growth (Kishimoto 2006). Studies have demonstrated that p53 is a transcriptional repressor of the IL6 promoter in cancer cell lines (Santhanam and others 1991; Angelo and others 2002), and using p53−/− mice, it has also been shown that p53 represses IL6 expression in the thymus following treatment with LPS (Komarova and others 2005). In this study, the finding that IL6 is highly abundant in p53−/− cardiac samples supports those previous studies showing that p53 also acts as the repressor of IL6 in the heart. Unexpectedly, IL6 was not highly abundant in the cardiac proteome of the p53−/−/STAT1−/− mice and IL6 mRNA was reduced in STAT1 −/− mice. These data suggest that STAT1 is an activator of IL6 expression in the heart.
High circulating IL6 levels are associated with cardiac dysfunction and heart failure and correlate with poor prognosis for patients having suffered myocardial infarction (Tsutamoto and others 1998; Kortekaas and others 2013). We observed STAT1-dependent gene regulation of IL6 in the heart. IL6 is known to be secreted from muscle cells (Song and others 2010) and has been dubbed a “myokine,” a term that was first used to describe muscle factors that are released upon exercise and thought to be responsible for potentiating the beneficial effect of exercise (Febbraio and Pedersen 2005). Muscle has also been shown to release myokines in pathological states, and consequently, to have systemic effects on energy metabolism and aging (Tyynismaa and others 2010; Durieux and others 2011; Suomalainen and others 2011). IL6 has pro- and anti-inflammatory effects, and thus, cardiac release of IL6 may contribute to the formation of atherosclerotic plaques. STAT1 is known be phosphorylated in the heart following ischemia, thereby inducing its dimerization and translocation to the nucleus, whereby it activates transcription of target genes (Stephanou and others 2000). Thus, regulation of IL6 by STAT1 is likely to be important for its secretion upon stress and its subsequent inflammatory effects.
IL6 signaling is mediated by the receptor-signaling component gp130, which is also a signal transducer for a number of other cytokines, including the strong hypertrophic cytokine cardiotrophin-1 (Liao and others 2002). Stimulation of the cardiac gp130 receptor leads to hypertrophy (Hirota and others 1995). Thus, IL6 secreted from cardiac myocytes may have auto- or paracrine functions, leading to stimulation of hypertrophic pathways in response to cardiac insufficiency, and eventually to a pathological hypertrophic response.
Interestingly, the increased IL6 expression observed in the cardiac tissue of p53−/− compared to p53−/−/STAT1−/− mice was not equaled in MEFs. Conversely, IL6 mRNA expression and secretion was higher in p53−/−/STAT1−/− compared to p53−/− MEFs, demonstrating the importance of tissue-specific regulation of IL6 by STAT1. Moreover, induction of IL6 protein expression by IFNγ treatment in p53−/−/STAT1−/− MEFs was not observed in p53−/− MEFs. It has previously been demonstrated that STAT1 is not required for IL6 production in MEFs treated with IFNγ (Samardzic and others 2001). IL6 has been implicated among the risk factors in inflammation-induced cancers (Rose-John and Schooltink 2007), and therefore, the increased IL6 expression in p53−/−/STAT1−/− mice might have contributed to spontaneous development of tumors that occur at a higher frequency, compared to p53−/− mice. However, the reason of higher IL6 expression in p53−/−/STAT1−/− MEFs compared to p53−/− MEFs remains to be determined.
Cardiovascular diseases are a major cause of morbidity and mortality worldwide. IL6 is an important cytokine in the etiology of cardiovascular diseases because of its role in inflammation and cardiac hypertrophy. We have described here a novel pathway that may be tissue specific in the heart, whereby regulation of the IL6 gene is differentially regulated by STAT1 and p53. Understanding the molecular mechanisms involved in the pathogenesis heart failure will lead to improved therapies and better treatment of patients.
Footnotes
Acknowledgments
The authors would like to thank Dr. Kevin Mills for assistance with mass spectrometry.
This work was funded by the British Heart Foundation and The Wellcome Trust.
Author Disclosure Statement
No competing financial interests exist.