
Abstract
Obesity is a world health problem that increases the risk for developing type 2 diabetes, cardiovascular disease, fatty liver, and some types of cancer. In postmenopausal women, it represents an important risk factor for the development of breast cancer (BC). Leptin is an adipokine that is secreted by fatty tissue, and high leptin levels are observed both in mouse models of obesity and in obese subjects. High levels of leptin promote the proliferation and progression of various types of cancer, including BC. This review provides a general overview of the biology of leptin, important laboratory studies, and animal and clinical models that have provided evidence for an active role of leptin in the proliferation, progression, and survival of mammary tumors. Finally, this review addresses the most recent studies on the use of leptin receptor antagonists as a novel therapeutic treatment for BC.
Introduction
O
AT is no longer simply considered an energy store. Instead, AT is an organ with a multiplicity of functions and is involved in processes related to energy metabolism, neuroendocrine function, and the secretion of peptides known as adipokines, which are proteins secreted by adipocytes. Similar to cytokines, adipokines can act in paracrine, autocrine, and endocrine manners in various physiological and metabolic processes (Kershaw and Flier 2004; Trayhurn and Wood 2004). Leptin is one of the most well-studied adipokines that participates in energy regulation as well as many other physiological functions. Leptin is secreted by white AT, and levels of this molecule are high in obese people. In vitro and clinical studies have implicated leptin in multiple obesity-related pathologies, such as BC. This report provides an overview of leptin biology and its role in the energy homeostasis, the principal in vitro studies undertaken thus far, and the animal and clinical models that have provided evidence for a role of leptin in BC progression.
A Little History
At the end of the 1940s, Ingalls and others (1950) identified a mutation in mouse (ob/ob) that gave them an obesity phenotype (Campfield and others 1995). The cloning and sequencing of the ob gene revealed a 167 amino-acid protein (leptin) with strong homology among vertebrates (Zhang and others 1995). Campfield and others (1995) first found a role for leptin in appetite control and energy balance. They observed that peripheral and central administration of recombinant leptin diminished food intake and the body weight of ob/ob mouse and mouse with diet-induced obesity but not of db/db mouse (Campfield and others 1995). Frederich and others (1995) suggested that leptin acted as a circulating signal involved in energy homeostasis.
Shortly after the discovery of leptin, Hummel and others (1966) found a mutation (db/db) in the obese mouse with metabolic abnormalities similar to those present in people with diabetes.
Intraspecific and interspecific mouse crosses segregating the db mutation, and it enables the genetic mapping of the db mutación (Bahary and others 1990). Since the autosomal recessive mutations fa (rat) and db (mouse) cause obesity syndromes that develop early and ultimately become severe, experiments and linkage data from Truett and others (1991) suggested that fa and db are mutations in homologous genes. The db/db mice did not respond to recombinant leptin administration, which suggested the presence of a defect in the receptor for the leptin signal, which could be codified by the db gene (Campfield and others 1995).
Tartaglia and Maffei found the leptin receptor (obR) in the choroid plexus and hypothalamus of mouse (Tartaglia and others 1995; Maffei and others 1995). High expression of mRNA for the obR was located in regions of the hypothalamus involved in the body weight regulation (Mercer and others 1996). Characterization of this protein showed that it was a trans-membrane receptor with features similar to those of class I family cytokine receptors (Tartaglia and others 1995). Chen and others (1996) confirmed that the absence of obR propitiates the obesity phenotype observed in db/db mouse and showed the importance of the long intracellular domain of the receptor (obRb) to mediate adequate intracellular signaling.
Getting to Know Leptin and the Receptor
After the discovery of leptin and the receptor, several subsequent studies clarified the structural and functional characteristics of this adipokine. Leptin is a 16 kDa secreted protein with a helical structure. This feature makes it a member of the class I family of cytokines (Madej and others 1995). The N-terminal region (94 amino acids) is important for interaction with the receptor and for the biological activity of leptin (Imagawa and others 1998). Adipocytes are the principal source of leptin mRNA (Frederich and others 1995). In humans, leptin mRNA expression occurs principally in mature adipocytes (Masuzaki and others 1995).
The obR (Fig 1) is a transmembrane protein with characteristics similar to those of the class I family of cytokine receptors, including IL-6, G-CSF, and LIF. The mouse and human forms of the obR have 78% and 71% homology, respectively, for the extracellular and intracellular domains (Tartaglia and others 1995). There are several isoforms of the obR in mouse and humans that arise from alternative RNA splicing at the most C-terminal coding exón: obRa, obRb, obRc, obRd, and obRe. All of them contain identical extracellular and transmembrane domains, and the differences between them lie in the cytoplasmic domain, which is of a different length and composition. The obRb isoforms contain a cytoplasmic domain with 302 amino-acid residues. The short isoforms (obRa, b and c) range in length from 32 to 40 amino acids. A common Box 1 motif is also found in the other 4 isoforms. The obRe isoform is a soluble receptor and does not contain a cytoplasmic domain (Lee and others 1996; Bjørbæk and others 1997; Tartaglia 1997). In humans, proteolytic breakages of the obRa and obRb isoforms, mediated principally by the proteases ADAM10 and to a lesser extent ADAM17, create the obRe isoform. The soluble form has been suggested to transport leptin from the periphery to its central place of action, the hypothalamus (Maamra and others 2001; Chen and others 2006).
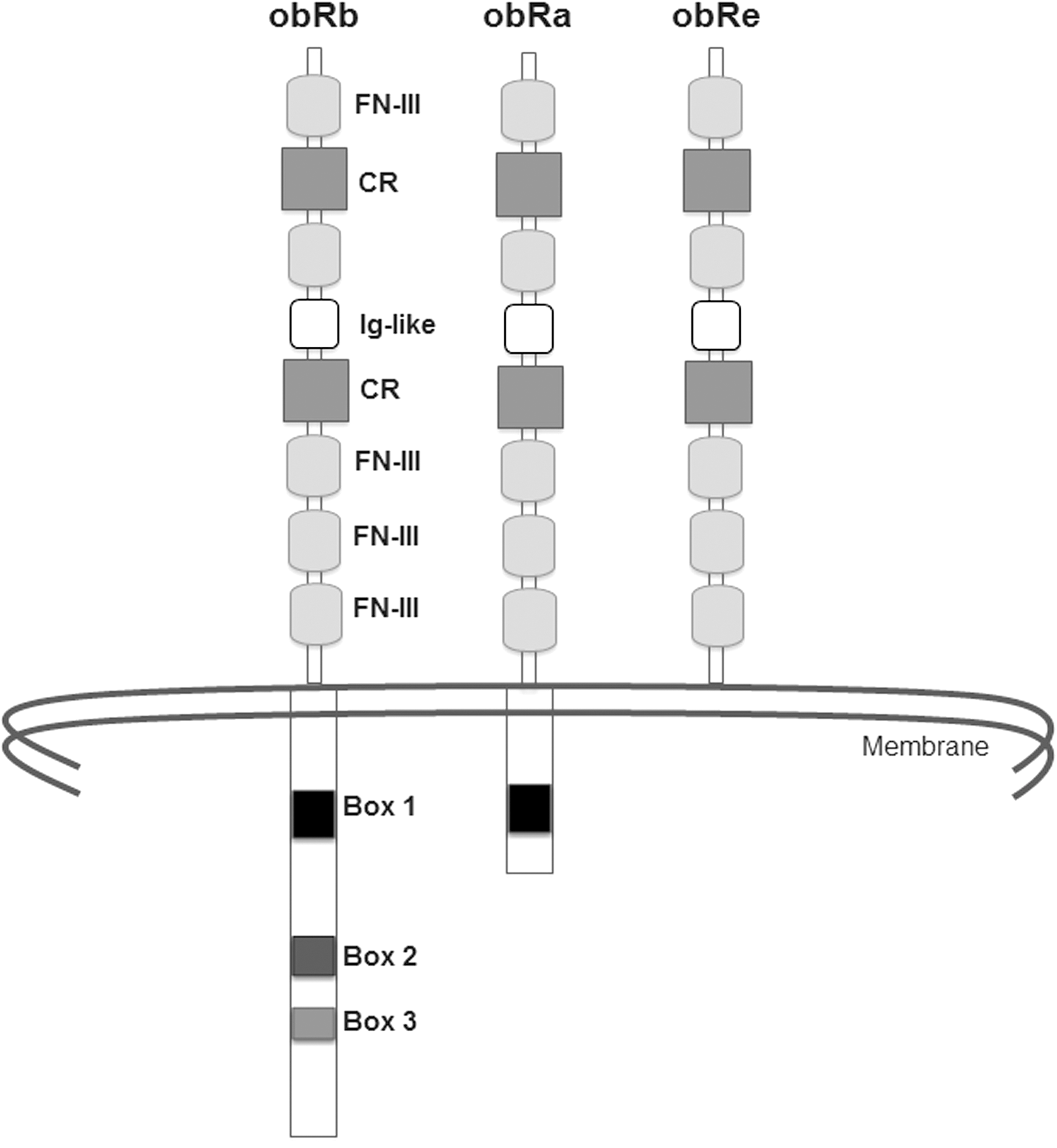
Structure and isoforms of leptin receptor. CR, citokine receptor homology; Ig-like, immunoglobulin-like fold; FN-III, fibronectin type III.
In the mouse, the obRb isoform is expressed principally in the arcuate, ventromedial, dorsomedial, and lateral nucleus of the hypothalamus. These areas are involved in regulating food intake and body weight (Fei and others 1997). The obRa isoform is highly expressed in the microvessels of the brain and in the choroidal plexus (Bjørbæk and others 1998). The obRa and obRb isoforms are expressed in peripheral tissues, including the adrenal glands, AT, heart, liver, lung, ovaries, pancreas, skeletal muscle, and testicles, although in some of these (adrenal glands, liver, pancreas, and testicles), the expression is less for obRb (De Matteis and others 1998).
Leptin is transported through the blood–brain barrier by a saturable system. This activity is attributed to the obRa isoform (Banks and others 1996; Golden and others 1997; Hileman and others 2002). Accordingly, Hileman and others (2002) showed that the obRa isoform mediates the transportation of leptin through polarized epithelial cells (Hileman and others 2000). The mechanism of entry by leptin into these cells occurs through clathrin-mediated endocytosis. Subsequently, leptin degradation occurs in a lysosomal compartment (Barr and others 1999; Uotani and others 1999).
The obRb isoform shares some functional characteristics with the IL-6 family of cytokine receptors. Typically, the signaling of these receptors is mediated by Janus kinase (JAK), signal transducer and activators of transcription (STAT), and mitogen-activated protein kinase (MAPK) (Heinrich and others 2003). At first, the obRb was believed to be the only isoform with a signaling capacity. Bjørbæk and others (1997) showed that the obRb and obRa isoforms activate leptin-mediated signaling pathways, such as phosphorylation of JAK2, Insulin receptor substrate 1 (IRS-1), and MAPK. However, only the obRb isoform has the capacity to activate STAT3 and to stimulate the expression of c-fox (Bjørbæk and others 1997).
Signaling Routes Mediated by Leptin
Leptin binding promotes dimerisation of the obRb chains (White and others 1997) and its subsequent association with and activation of JAK2. Once activated, Jak2 phosphorylates the tyrosine residues (conserved in mouse and humans) Tyr985, Tyr1077, and Tyr1138 from the obRb cytoplasmic domain. This recruits other downstream signaling molecules, such as STAT3/5/6 (Baumann and others 1996; Ghilardi and others 1996). Tyr1138 phosphorylation residue allows STAT3 to be recruited and phosphorylated.
The translocation of STAT3 to the nucleus mediates the transcription of target genes, including pro-opiomelanocortin (POMC), agouti-related protein (AgRP), and suppressor of cytokine signaling 3 (SOCS-3), a negative regulator that inhibits obRb/JAK2 signaling (Banks and others 2000). The Tyr985 residue is also a binding site for SHP2 (protein-tyrosine phosphatase). The leptin interaction with obRb induces its own phosphorylation and direct association with the leptin receptor. SHP2 activation leads to diminished JAK2 phosphorylation, suggesting that it is most likely involved in attenuating the leptin-mediated signaling (Li and Friedman 1999). SHP2 recruitment also promotes growth factor receptor-bound protein 2 (GRB2) binding, a necessary event for extracellular signal-regulated kinases (ERK) activation and the accumulation of c-fos mRNA (Banks and others 2000).
There are also other intracelular signalling pathways that are involved in the activation of the leptin receptor. Leptin lightly stimulates phosphatidylinositol kinase-3 (PI3K) that is associated with IRS-1 in tissues sensitive to insulin (Kim and others 2000). Hill and others (2008) showed that leptin activates POMC neurons through a PI3K-dependent mechanism (Hill and others 2008). Cota and others (2006) demonstrated that leptin increases the activation of hypothalamic protein mammalian Target of Rapamycin (mTOR), a necessary step for the appetite-suppressing activity of leptin (Cota and others 2006) (Fig 2).
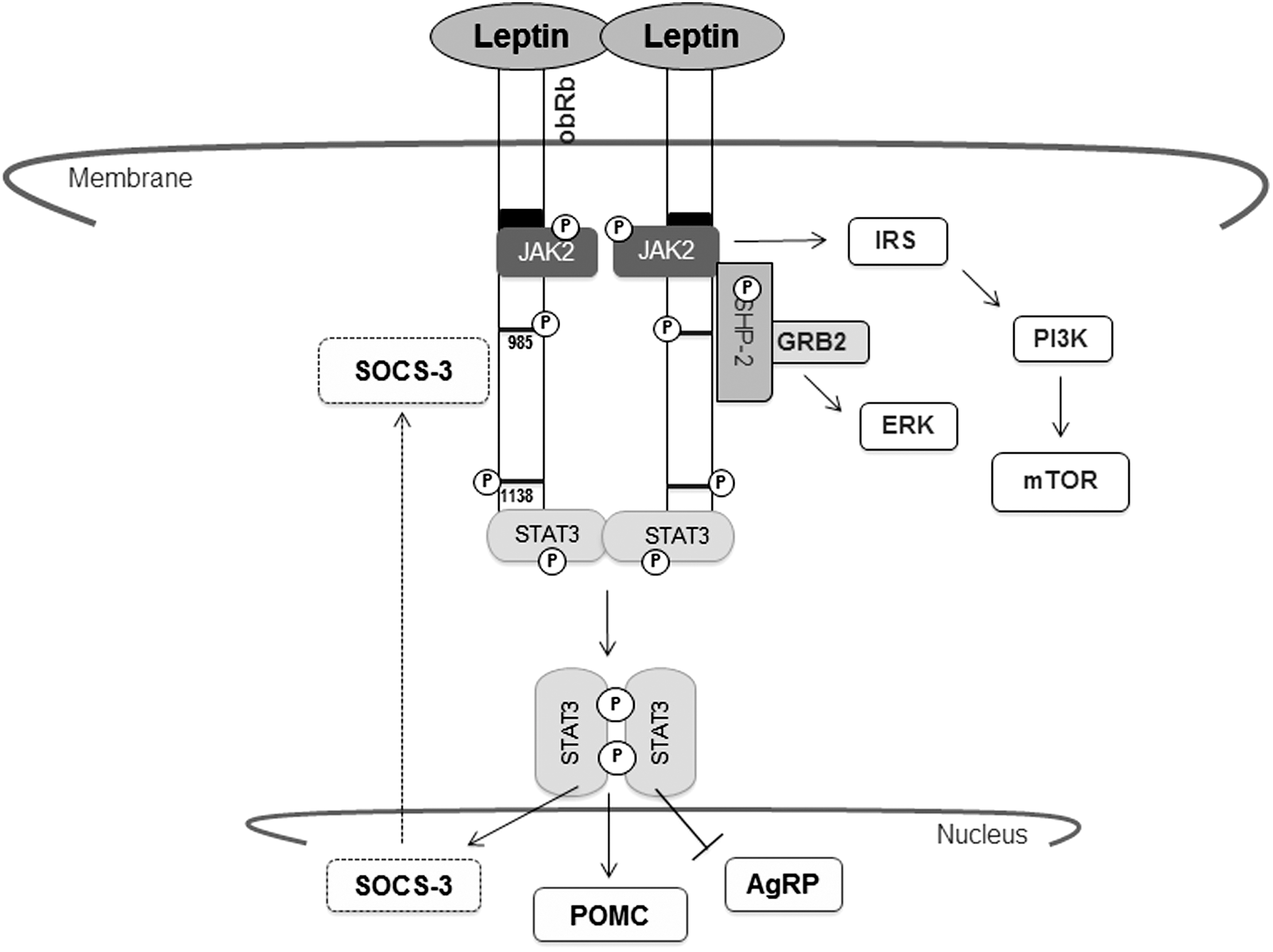
Leptin-induced signaling pathways. JAK2, janus kinase 2; STAT3, signal transducer and activators of transcription 3; POMC, pro-opiomelanocortin; AgRP, agouti-related protein; SOCS-3, suppressor of cytokine signaling 3; SHP2, protein-tyrosine phosphatase; GRB2, growth factor receptor-bound protein 2; ERK, extracellular signal-regulated kinases; IRS-1, insulin receptor substrate 1; PI3K, phosphatidylinositol kinase-3; mTOR, protein mammalian target of Rapamycin.
Energy Homeostasis
Leptin represents a physiological signal that controls energy balance. To do so, specific neuropeptides are activated or repressed by leptin. The neuropeptide Y (NPY) and POMC neurons are located in the ARC nucleus of the hypothalamus, and both of them express the obRb isoform (Schwartz and others 1996). The anorexigenic effects of leptin are mediated by the proteolytic processing of POMC and the subsequent liberation of ∝MSH (melanocyte-stimulating hormones), an important anorectic mediator. The activation of the pomc gene is dependent on STAT3 activation. The antagonist neuropeptides AgRP and NPY are also expressed in the ARC nucleus, and both are negatively regulated by leptin (Münzberg and others 2003).
The Other Side of Leptin
Obesity represents an important risk factor for BC in postmenopausal women (Sonnenschein and others 1999). Levels of leptin are often high in mouse models of obesity and in obese humans (Frederich and others 1995; Lonnqvist and others 1995; Maffei and others 1995; Considine and others 1996). Recently, an active role for leptin in the initiation and progression of BC has been recognized. The precise mechanism is not yet completely understood, but several in vitro studies and animal and clinical models have provided some clues to the participation of leptin in the development of BC.
In Vitro Studies (Table 1)
Cell proliferation
Leptin promotes the proliferation of both normal and malignant epithelial cells. Obese mice deficient in leptin or its receptor present abnormal development of the mammary gland and do not develop tumors (Hu and others 2002; Cleary and others 2004). The proliferative effect of leptin is mediated by the activation of different signaling pathways. In BC cell lines, leptin promotes the activation of STAT3, ERK, and activator protein 1 (AP-1) (Hu and others 2002) and induces the expression of c-myc and steroid receptor coactivator-1 (SCR-1), a coactivator of the p160 family (Yin and others 2004). In the MCF7 cell line, leptin up-regulates the expression of aromatase mRNA and its enzymatic activity. The regulation of the aromatase promoters is modulated by AP-1 (Catalano and others 2003).
The receptor HER2/Neu (ErbB2) belongs to the epidermal growth receptor family and is overexpressed in 20%–30% of cases of BC and cervix cancer (Nair 2005). Leptin mediates the transactivation of HER2 and subsequent cell proliferation in the SKBR3 cell line (Soma and others 2008). In the MCF7 line, as in BC tissues, it was observed that HER2 colocalizes with obR, which suggests the possibility of interaction between these molecules (Fiorio and others 2008). High levels of glucose and leptin also increase the proliferation of MCF7 cells (Okumura and others 2002).
Leptin positively regulates the promoter of the Cyclin D1 gene in the MCF7 cell line. Coactivators, histone acetyltransferases, SRC1, and mediator complex subunit 1 (Med) are recruited to this promoter and subsequently promote the overexpression of cyclin D1 (Saxena and others 2007).
Furthermore, leptin induces the genes cyclin A2, G, and cyclin-dependent kinase 2 (CDK2), CDKN1A(p21), and CDKN2A(p16); while it reduces the genes involved in the inhibition of the G1 cell-cycle phase cyclin-dependent kinase inhibitor CDKN1B(p27). This confirms leptin's participation in cell cycle progression (Perera and others 2008a).
Survival
A number of studies have shown that antiapoptotic effects can be mediated by leptin. In human umbilical vein endothelial cells (HUVECs) and human adult micro and macrovascular endothelial cells, leptin mediates the over-regulation of B-cell lymphoma 2 (bcl-2), an inhibitor of apoptosis (Artwohl and others 2002). Leptin induces the expression of Survivin and Hey2, a pro-survival factor and a repressor, respectively, both of which are Notch genes. Apart from inducing the expression of the anti-apoptotic genes bcl2 and survivin, leptin reduces the expression of interacting protein (TRAIP), insulin-like growth factor 1 receptor (IGF1R), and tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD) (Perera and others 2008a).
In the MCF7 line, leptin promotes activation of the protein kinase B/glycogen synthase kinase 3 (Akt/GSK3) antiapoptotic signaling pathway as well as the phosphorylation of retinoblastoma protein. The exposure of cells to 10 nM ICI 182,780 (7-alkylsulfinyl analog of the endogenous estrogen 17-estradiol and binds to both estrogen receptor (ER) subtypes with a comparable affinity to 17-estradiol) blocked cell proliferation, induced rapid ER degradation, inhibited nuclear ER expression, and reduced ER-dependent transcription. The antiestrogenic activity of ICI 182,780 is attenuated in this same cell line by simultaneous treatment of cells with 100 ng/mL leptin (Garofalo and others 2004).
Progression and invasion
The usage of protease techniques allowed Perera and others (2008b) to identify a series of proteins regulated by leptin and secreted by the MCF7 cell line. Among these proteins are a precursor of collagen, KF10, the isoform Cortactin, and NY-BR-62 (serologically defined BC antigen). These observations suggest that leptin alters the tumor microenvironment to favor its growth and progression. In addition, an increase was observed with fibroblast growth factor 9 (FGF-9) that was accompanied by a reduction in the levels of insulin-like growth factor binding protein-2 (IGFBP-2) and transforming growth factor-ß3 (TGF-ß3). The autocrine/paracrine effects of these growth factors can also contribute to the proliferation, migration, and invasion of the tumor (Perera and others 2008a).
FGFs regulate cellular processes such as proliferation, survival, migration, and differentiation. Alteration in FGR signaling may favor the growth of the tumorous cells (Turner and Grose 2010). IGF-1 and II are powerful mitogens that act in a synergistic manner to stimulate the growth of BC cells. IGFBPs regulate the biodisposition and action of the IGFs (Sachdev and Yee 2001); thus, reduced levels of IGFBP3 can favor the proliferative effects of IGF-1 in the tumorous cells (Perera and others 2008a). TGF-ß is a growth inhibitor of mammary tissue epithelial cells. In the early stages of tumor genesis, it is a tumor suppressor of tumors. However, in later stages, TGF-ß can promote tumor progression and metastasis in bones and lungs (Drabsch and Dijke 2011).
The matrix metalloproteinases (MMPs) are enzymes involved in the invasion and metastasis of tumors. MMP1, MMP2, and MMP9 are highly expressed in human BC tissue (Iwata and others 1996). Leptin promotes the invasion of MCF7 cells through increased activity of MMP2, which is dependent on c-Jun N-terminal kinases (JNK) (McMurtry and others 2009). E-caderine is an intercellular adhesion molecule related to epithelial tumors. In 3-dimensional cultures of MCF-7 cells, leptin promotes proliferation and cellular adhesion-dependent expression of E-caderine (Mauro and others 2007).
Leptin mediates the overexpression of extracellular matrix genes, including CTCF (connective tissue growth factor), villin 2, and basigin. These genes have been associated with BC progression and metastasis (Perera and others 2008a).
The epithelial–mesenchymal transition (EMT) consists of a reversible change in the phenotype of epithelial cells and is principally related to the stage of development, tissue remodeling, and wound repair. A process similar to EMT is observed during the progression and metastasis of tumors (Savagner 2010). An important step during EMT is the activation of ß-catenine, a protein involved in the Wnt (wingless) signalling.
In the absence of a linking Wnt signal, the ß-catenin destruction complex catalyzes ß-catenin phosphorylation and its subsequent degradation in the proteasome. The presence of a Wnt signal inhibits ß-catenin phosphorylation. Accumulation of this protein in the cytosol and its translocation to the nucleus enables transcription of white Wnt genes (Xu and Kimelman 2007).
Recently, Yan and others (2012) have shown that leptin induces EMT in BC cells, a new and previously undescribed function. MCF7 cells treated with leptin exhibit a phenotype similar to EMT. Mesenchymal markers such as vimentine, N-caderine, and fibronectine were also up-regulated. Leptin induces ß-catenin activation and nuclear translocation, which is modulated by the Akt/GSK3ß and MTA/Wnt pathways. Furthermore, leptin-mediated GSK3ß phosphorylation allowed disassociation of the ß-catenin destruction complex. RT-PCR and western blotting were used to show overexpression of cyclin D1 and fibronectin, genes that induce EMT (Yan and others 2012).
Angiogenesis
There is evidence supporting leptin-mediated promotion of angiogenesis. In 4T1 mouse mammary cancer cells, leptin treatment increases the expression of vascular endothelial growth factor (VEGF) and VEGFR2 (Gonzalez and others 2006). VEGF regulation is mediated by the activation of hypoxia inducible factor-1 (HIF-1α) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (Gonzalez-Perez and others 2010). In 4T1 cells, leptin activates various signaling pathways to mediate activation of the IL-1 system (Zhou and others 2011).
Notch-mediated signaling participates in several physiological and development processes (Bray 2006). In mouse MCF7 xenografts, Notch promotes angiogenesis (Soares and others 2004). Gonzalez-Perez and others (2010) showed that leptin is a regulator of Notch expression and activation. The induction of VEGF/VEGFR2 is dependent on Notch-IL-1-Leptin crosstalk (NILCO) in BC cells (Guo and Gonzalez-Perez 2011). In vitro studies have showed that leptin-mediated signaling activates mechanisms that promote the progression of the tumor development.
Studies in Animal Models
The role of leptin in the physiopathology of BC in animals turns out to be more complex than was suggested by studies conducted in vitro. Rodent models are frequently used in the study of BC, as they easily develop spontaneous or induced tumors (Szpirer and Szpirer 2007).
However, leptin deficiente obese Lep ob Lep ob mice have reduced incidences of spontaneous and oncogene-induced mammary tumors (Cleary and others 2003). In addition, no mammary tumors were detected in MMTV-TGF-α/Lep db Lep db mice; whereas the incidence of mammary tumors in MMTV-TGF-α/Lep+Lep + mice and MMTV-TGF-α/Lep + Lep db mice was 69% and 62%, respectively. Despite MMTV-TGF-α/Lep db Lep db mice having elevated serum leptin levels, mammary tumor development did not occur (Cleary and others 2004).
By contrast, Park and others (2010) showed using a mouse model system for peripheral leptin receptor deficiency (db/dbNse/Nse) in MMTV-PyMT mouse that reconstitution of obRb specifically in neurons allowed for re-establishment of epithelial mammary duct formation. In this model of peripheral obRb deficiency, a significant reduction was observed in the growth of the primary tumor and metastasis to the lung accompanied by decreased ERK1/2 and STAT3 signaling. The tumor cells deficient in obRb also exhibited greater mitochondrial respiration. Based on these findings, the authors suggested that a microenvironment with high local concentrations of leptin and subsequent stimulation of obRb mediates the activation of ERK1/2 and STAT3, routes that promote proliferation, survival, and metastasis of the tumor. At the same time, cellular metabolism predominates where the Warburg effect is used as the principal source of energy (Park and others 2010).
Zheng and others (2011) suggested that a functional obR is required in tumorous cells and that an environment with excess leptin promotes the development of the tumor. Using tumorous cells derived from a transgenic MMTV-Wnt-1 mouse that were transplanted into WT, db/db, and ob/ob mouse, leptin deficiency was found to suppress the growth of mammary, and phosphorylated Akt (p-Akt) was also dependent on leptin (Zheng and others 2011). These data suggest that both leptin and an intact leptin-signaling pathway are necessary for normal mammary gland development and for mammary gland tumorigenesis in vivo (Hu and others 2002), and elevated serum leptin levels may enhance cell signaling and promote proliferation (Cleary and others 2004).
Clinical Studies
Obesity is a risk factor for BC in postmenopausal women (Rose and Vona-Davis 2010). Metabolic syndrome is frequently seen in obese people. Individual components (as insulin resistance, aromatase activity, adipokine production, angiogenesis, glucose utilization, and oxidative stress/DNA damage) of the metabolic syndrome contribute to the development and progression of cancer via a systemic and/or local mechanism (Cowey and Hardy 2006).
An active role for leptin in the initiation and progression of BC can be achieved through endocrine, paracrine, or autocrine mechanisms. For example, serum from patients with BC have shown elevated serum leptin levels with regard to controls (endocrine pathway) (Chen and others 2006; Wu and others 2009). On the other hand, the breast microenvironment is influenced by adipocyte, stromal cells, and factors (Iyengar and others 2003). It has been shown that leptin is produced by preadipocytes on TNF-alpha or IL-1beta stimulation.
Stimulus related to obesity, such as insulin, IGF-1, oestrogens, and hipoxic conditions, can also contribute to increases in this adipokine. These data suggest that adipokines produced locally in mammary tissue act in a paracrine pathway, and such interactions can influence cancer progression (Simons and others 2005; Garofalo and others 2006; Carroll and others 2011). Finally, there is an overexpression of leptin and the receptor in breast tumor tissue, and it has been associated with a poor prognosis (Table 2). These data suggest that leptin may indeed have an autocrine role in mediating progression of BC. Taking the studies mentioned earlier into account, it might be beneficial to quantify the levels of leptin in tissue or serum as a diagnostic and/or prognostic biomarker of BC.
Leptin as a New Therapeutic Target for BC
In vitro and animal model studies have clearly shown a role for leptin and its receptor in the development and progression of BC. Modulating the favorable effects of leptin could help in treating illnesses related to obesity and cancer. A number of researchers have begun studying leptin antagonists as possible therapeutic treatments.
Gonzalez and others (2009) evaluated the effect of treatment with the PEGylated antagonist of the LEPR (PEG-LPrA2) in mice with xenoinserts of MCF-7 (RE+) and MDA-MB231 (RE−) cell lines. Tumor swelling diminished considerably in the RE+ tumors (>40 times) with regard to RE− tumors (2 times). Using immunohistochemistry, a significant reduction was shown in the levels of leptin, LEPR, VEGF, VEGFE2, PCNA, IL-1R, and cycline D1 in RE+ tumors of mice treated with PEG-LPrA2. These effects were also observed in RE− tumors; however, they were more evident in RE+ tumors (Gonzalez and others 2009). Ferla and others (2011) found that treatment with the LEPR antagonist Aca1 at 10 to 25 nN suppressed the formation of tubes induced by leptin and the mitogenesis of HUVEC, respectively (Ferla and others 2011). Recently, Otvos and others (2011) evaluated the effects of the antagonist Allo-aca in a BC triple-negative cell line (MDA-MB-231) model. The findings indicated that treatment with Allo-aca inhibited leptin-induced proliferation at a concentration of 50 pM. Moreover, in the xenoinsert orthotopic mouse model using the MDA-MB-231 cell line, subcutaneous administration of Allo-aca significantly prolonged survival time from 15.4 days (controls with no treatment) to 24 days (dose 0.1 mg/Kg/day) and 28 days (dose 1 mg/Kg/day) (Otvos and others 2011). Using LEPR as a therapeutic target represents a new alternative treatment for BC (Otvos and Surmacz 2011). LEPR overexpression has been observed in BC and other types of cancer, and while the use of LEPR antagonists as a part of the treatment for BC still requires extensive research, the preliminary findings thus far are promising, and the antagonists might even be effective against other types of cancer, especially those related to obesity.
Conclusion
Obesity is a condition that provokes the development of several pathologies, with BC among them. Leptin is an adipokine secreted by AT, and levels of leptin are found to be high in animal models of obesity as well as in obese humans. The information presented in this article demonstrates the central role played by leptin in provoking the proliferation, progression, and survival of tumor mammary tissue. However, further research is needed to understand the molecular bases of these effects in greater depth, as they may represent new therapeutic targets in the treatment of BC.
Footnotes
Author Disclosure Statement
No competing financial interests exist.