
Abstract
Tripartite motif (TRIM) 22 plays an important role in interferons (IFNs)-mediated antiviral activity. We previously demonstrated that interferon regulatory factor-1 (IRF-1) played a central role in IFN-γ-induced TRIM22 expression via binding to a special cis-element named 5′ extended IFN-stimulating response element (5′eISRE). In this study, we sought to identify the signaling pathways involved in TRIM22 induction by IFN-γ. By using various pharmacological inhibitors, it was found that the activity of tyrosine kinase and phosphatidylcholine-phospholipase C (PC-PLC), but not phosphatidylinositol-phospholipase C (PI-PLC) and phospholipase D (PLD), was required for IFN-γ-induced TRIM22 expression in HepG2 cells. Tyrosine kinase Janus kinase (JAK), not SRC and PYK2, played an indispensable role in TRIM22 induction. Inhibition of protein kinase C (PKC) activity also significantly attenuated IFN-γ induction of TRIM22. Although treatment with IFN-γ resulted in the stimulation of mitogen-activated protein kinases (MAPKs) (p38, ERK, and JNK) and pI3K/Akt/mTOR pathways in HepG2 cells, the inhibition of their activity did not affect IFN-γ-stimulated TRIM22 expression. Further studies showed that overexpression of JAK1 and PKCα activated TRIM22 promoter activity in a 5′eISRE-dependent manner, and inhibition of not only JAK but also PC-PLC/PKC pathways significantly attenuated IFN-γ-induced IRF-1 expression in HepG2 cells. Taken together, these data indicated that IFN-γ induced TRIM22 expression via activation of JAK and PC-PLC/PKC signaling pathways, which involved the cis-element 5′eISRE and the transactivator IRF-1.
Introduction
T
With the aim of elucidating the molecular mechanisms of TRIM22 induction by IFNs, we previously identified a special cis-element named 5′ extended IFN-stimulating response element (5′eISRE), which was crucial for IFN-γ-induced TRIM22 expression, and demonstrated that interferon regulatory factor-1 (IRF-1) played a central role in TRIM22 induction via binding to this cis-element. Furthermore, the association of IRF-1 with 5′eISRE was also revealed to be important for TRIM22 induction by IFN-α, as well as for basal TRIM22 expression (Gao and others 2010). Our more recent investigation further demonstrated that chromatin-remodeling enzyme BRG-1 played a critical role in TRIM22 induction by IFN-γ via controlling IRF-1 recruitment (Wang and others 2011). However, little is known about the signaling pathway involved in IFN-γ-induced TRIM22 expression.
One of the main signaling pathways activated by IFN-γ involves sequential phosphorylation of the tyrosine residues of the Janus kinase (JAK) and signal transducer and activator of transcription (STAT) proteins. STATs are then dimerized and translocated into the nucleus where they regulate gene expression. However, evidence indicates that the responses are more complicated (Platanias 2005; Stark 2007; van Boxel-Dezaire and Stark 2007). In addition to JAK-STAT pathway, the transcriptional response to IFN-γ may also involve phospholipases (Sands and others 1994; Chang and others 2002; Tsai and others 2009). There are 3 types of phospholipases: phosphatidylinositol-phospholipase C (PI-PLC), phosphatidylcholine-phospholipase C (PC-PLC), and PC-phospholipase D (PC-PLD). Activation of PLC and PLD can result in the activation of protein kinase C (PKC) via stimulating the generation of diacylglycerol (DAG) directly or indirectly, thus contributing to the induction of interferon-stimulated genes (ISGs). There has been also some evidence implicating mitogen-activated protein (MAP) kinases (p38, ERK, and JNK) in the induction of IFN responses (Katsoulidis and others 2005; Li and others 2010; Park and others 2011). Furthermore, the phosphatidylinositol 3-kinase (pI3K) is revealed to regulate transcriptional activation by IFN, whereas the Akt/mammalian target of rapamycin (mTOR) pathway plays an important role downstream of pI3K, promoting mRNA translation of ISGs (Kaur and others 2005; Fang and others 2006; Venkatesan and others 2006; Kaur and others 2007).
In the present investigation, we found that besides the classical JAK pathway, the PC-PLC/PKC pathway also played an important role in IFN-γ induction of TRIM22; while other signaling pathways, such as mitogen-activated protein kinases (MAPKs) and pI3K/Akt/mTOR, appeared to be not involved in this event. Further studies showed that JAK1-, PKC-α-mediated TRIM22 promoter activity was dependent on 5′eISRE, and JAK and PC-PLC/PKC pathways were revealed to be important for IFN-γ-induced IRF-1 expression.
Materials and Methods
Materials
Antibodies against TRIM22 were described in our previous study (Gao and others 2009; Gao and others 2010). Antibodies against phospho-/total form of p38, ERK, JNK, AKT, and p70s6k were purchased from Cell Signaling Technology (Beverley, MA). Antibodies against STAT1, IRF-1, phospho-/total form of PKCα, and Actin were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Human recombinant IFN-γ was purchased from PeproTech (Rocky Hill, NJ). Genestine, daidzein, tyrphostin AG126, U73122, D609, JAK inhibitor I, PP2, Ro 31-8220, SB203580, U0126, SP600125, LY294002, tyrphostin A9, and Akt inhibitor (1L-6-Hydroxymethyl-chiro-inositol2-[(R)-2-O-methyl-3-O-octadecylcarbonate]) were obtained from Calbiochem (San Diego, CA). Propranolol and staurosporine were purchased from Sigma (St. Louis, MO).
Quantitative RT-PCR
Extraction of total RNA, reverse transcription were carried out as previously described (Gao and others 2009; Gao and others 2010). cDNA was subjected to real-time PCR in a 20 μL reaction mixture with the following primers: TRIM22, forward, 5′-ACCAAACATTCCGCATAAAC-3′ and reverse, 5′-GTCCAGCACATTCACCTCAC-3′; IRF-1, forward, 5′-TAT CGAGGAGGTGAAAGACC-3′ and reverse, 5′-TGCATGTAGCCTGGAACTG-3′; GAPDH, forward, 5′-ATCCCATCACCATCTTCCAG-3′ and reverse, 5′-GA GTCCTTCCACGATACCAA-3′. TRIM22 or IRF-1 expression was calculated following normalization to GAPDH levels by the comparative ΔΔ threshold cycle method. The specificity of the amplification reactions was confirmed by melt curve analysis. Results were representative of 3 independent experiments.
Western blotting
Protein was denatured in SDS, electrophoresed on SDS-PAGE (8% or 10% gel), and transferred onto a polyvinylidene difluoride microporous membrane (Millipore, Billerica, MA). Non-specific binding was blocked with TBST (50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 0.1% Tween 20) containing 5% (w/v) non-fat milk for 2 h at room temperature. After overnight incubation at 4°C with the indicated antibody, membranes were washed thrice with TBST and incubated at room temperature for 2 h with the corresponding horseradish peroxidase-conjugated secondary antibody diluted 1:1,000 in blocking buffers. After washing, signals were detected by an enhanced chemiluminescence kit (Pierce, Rockford, IL).
Transient expression and the promoter reporter assay
To determine the effect of pharmacologic inhibitors on IFN-γ-induced TRIM22 promoter activity, HepG2 cells were transfected with a TRIM22 promoter-dependent luciferase reporter plasmid (pLUC-160) (Gao and others 2010). At 24 h posttransfection, HepG2 cells were preincubated with various inhibitors for 30 min and then treated with IFN-γ for another 24 h, followed by detection of the luciferase activity in the cell lysates using the Luciferase Reporter Assay System (Promega, Madison, WI). In all transfection assays, pCMV-β-gal was cotransfected to normalize the transfection efficiency. Results were representative of 3 independent experiments.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Gao and others 2010). In brief, after cross-linking with formaldehyde, HepG2 cells were resuspended in cell lysis buffer and nuclei were centrifuged at 2,500 rpm for 5 min. The nuclei were isolated and sonicated on ice to shear the DNA to 200–1,000 bp. Chromatin was immunoprecipitated from 200 μL aliquots at 4°C by mild agitation overnight with 5 μg of a rabbit-specific antibody against STAT1, and the immune complexes were collected by incubation with protein A/G agarose (Upstate Biotechnology, Lake Placid, NY). To analyze the target region, the immunoprecipitated chromatin DNA samples were amplified by real-time PCR. The sequences of IRF-1 promoter primer are as follows: forward, 5′-CGCCAGGAGGGT GAAAAGATG-3′ and reverse, 5′-GGCTGCGTGCCGTCAT TTCG-3′. Data are presented as fold differences relative to input and values obtained by normal rabbit IgG using the formula 2[(CtIgG−Ct Input)−(CtAb−CtInput)], where Ct is the threshold cycles, IgG is the normal rabbit IgG, Ab is the specific antibody, and Input is the input genomic DNA.
Short interference RNA assay
Transfection of short interference RNAs (siRNAs) targeting IRF-1 or PKCα was performed as previously described (Gao and others 2010; Wang and others 2011).
Statistics
Results were reported as means±SD. T test was applied to comparisons between groups; a P value <0.05 was considered statistically significant.
Results
Role of tyrosine kinase, phospholipase C, and phospholipase D in IFN-γ-induced TRIM22 expression
To examine whether tyrosine kinase activation was involved in IFN-γ-induced TRIM22 expression in HepG2 cells, tyrosine kinase inhibitors genestine and tyrphostin AG126 were used. Pretreatment of cells for 30 min with genistein or tyrphostin AG126 inhibited the IFN-γ induction of the TRIM22 gene at both the mRNA and protein levels; while daidzein, an inactive analog of genistein, exerted little effect (Fig. 1A, B), indicating the implication of tyrosine kinase in TRIM22 induction by IFN-γ.
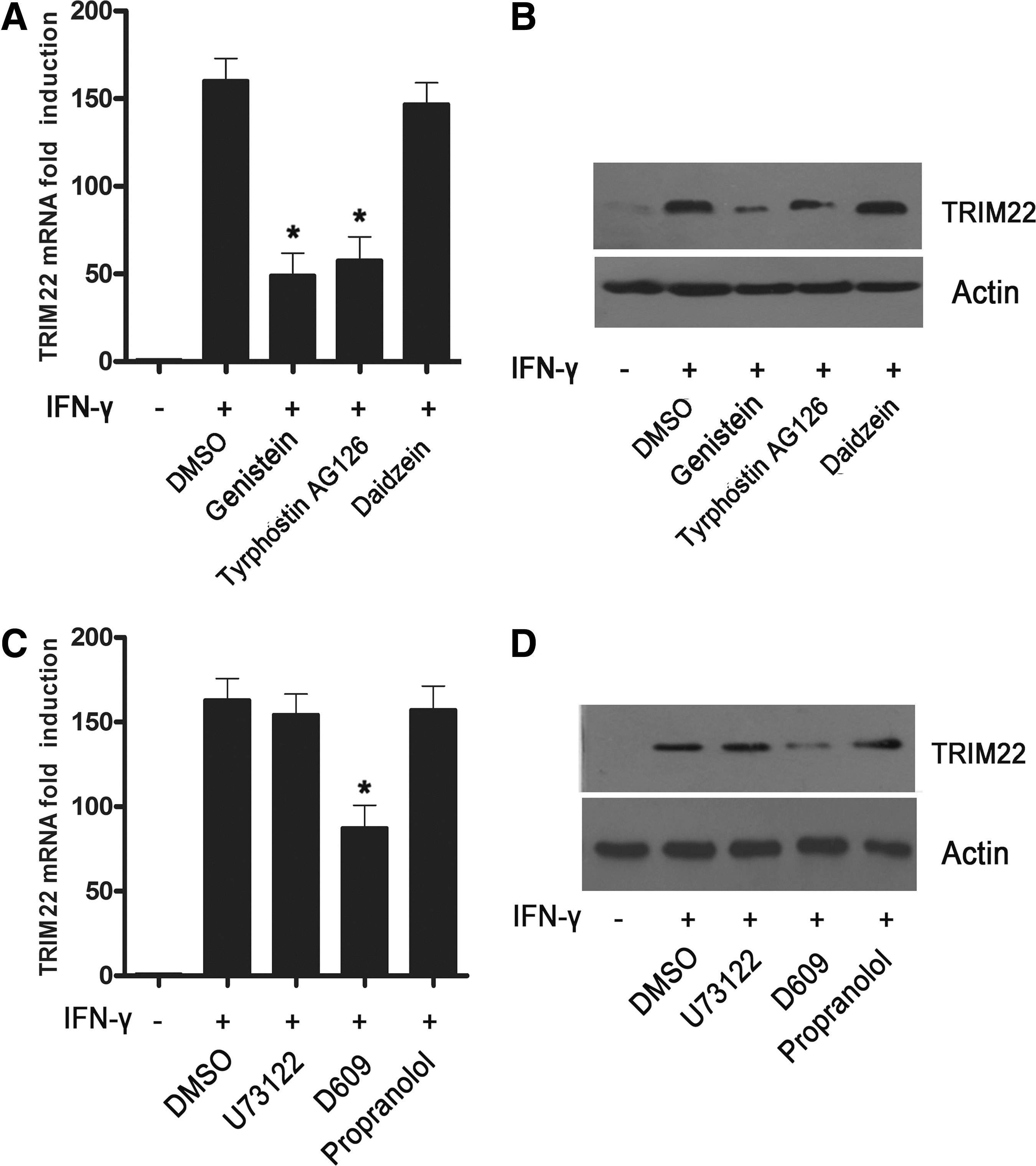
Role of tyrosine kinase and phospholipases in IFN-γ-induced TRIM22 expression.
To investigate the role of phospholipases in TRIM22 induction by IFN-γ, pharmacological inhibitors U-73122, D609, and propranolol were used. U73122 and D609 selectively inhibited the activity of PI-PLC and PC-PLC, respectively. Propranolol is the inhibitor of phosphatidate phosphohydrolase, which is required for PC-PLD-derived DAG formation (Nishizuka 1995; Brindley and others 2009). As shown in Fig. 1C and D, IFN-γ-induced TRIM22 mRNA and protein level was inhibited by D609, but not by U73122 and propranolol, indicating that PC-PLC, not PI-PLC or PC-PLD, was involved in IFN-γ induction of TRIM22.
Role of tyrosine kinase JAK, PYK2, or SRC in IFN-γ-induced TRIM22 expression
The data provided earlier showed that the activity of tyrosine kinase was required for IFN-γ-induced TRIM22 expression. Evidence indicates that, besides JAK, tyrosine kinases PYK2 and SRC also play important roles in ISGs induction by IFN-γ (Takaoka and others 1999; van Boxel-Dezaire and Stark 2007; Tsai and others 2009). To further investigate the role of tyrosine kinases in TRIM22 induction by IFN-γ, we treated HepG2 cells with JAK inhibitor I (a pan JAK inhibitor), tyrphostin A9 (a PYK2 inhibitor), and PP2 (a SRC inhibitor), respectively, and then tested TRIM22 expression induced by IFN-γ. Results showed that JAK inhibitor I nearly led to the complete block of TRIM22 expression, while tyrphostin A9 and PP2 had little effect, had no such effect (Fig. 2A, B), indicating that IFN-γ-induced TRIM22 expression was via the activation of tyrosine kinase JAK, not PYK2 and SRC.
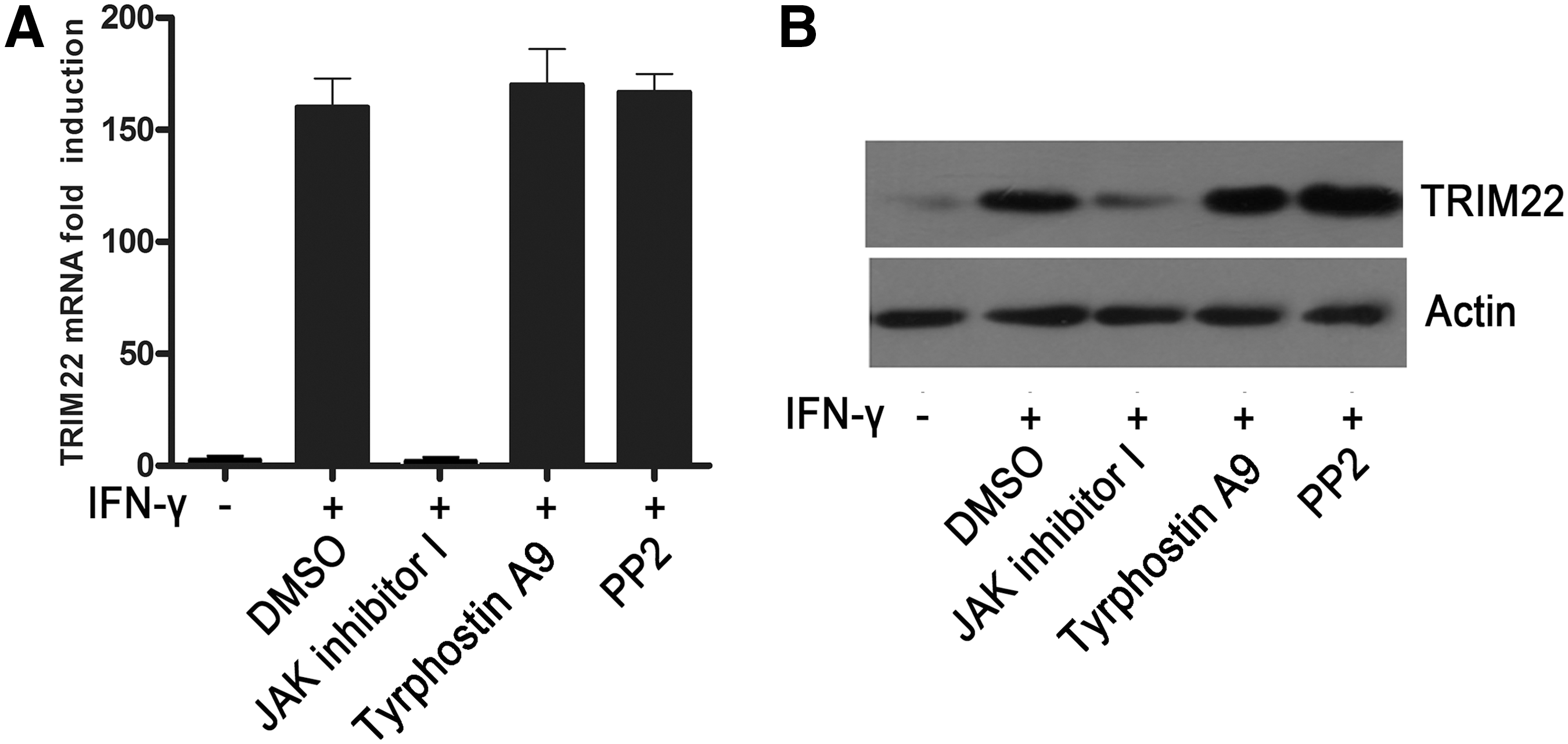
Role of tyrosine kinase JAK, PYK2, or SRC in IFN-γ-induced TRIM22 expression. HepG2 cells were pretreated with JAK inhibitor I (1 μM), tyrphostin A9 (1 μM), and PP2 (50 μM) for 30 min, respectively, and then tested the effect of IFN-γ (1,000 U/mL) on TRIM22 expression.
Role of PKC in IFN-γ-induced TRIM22 expression
The inhibition of IFN-induced TRIM22 expression by D609 implies the involvement of the PC-PLC pathway in this response. Activation of PC-PLC increases the generation of DAG and then activates PKC (Brindley and others 2009). To investigate the role of PKC in IFN-γ-induced TRIM22 expression in HepG2 cells, 2 PKC inhibitors, staurosporine and Ro 31-8220, were used. As shown in Fig 3A and B, IFN-γ-induced TRIM22 expression was significantly repressed by both staurosporine and Ro 31-8220, indicating the possible role of PKC activation in IFN-γ-mediated TRIM22 expression.

Role of PKC in IFN-γ-induced TRIM22 expression. HepG2 cells were pretreated with PKC inhibitors Ro 31-8220 (2 μM) and staurosporine (100 nM), respectively, for 30 min, followed by the treatment of IFN-γ (1,000 U/mL) for 24 h.
IFN-γ induces the activation of p38, ERK, and JNK MAPK in HepG2 cells, and lack of inhibition by SB 203580, U0126, and SP600125 in IFN-γ-induced TRIM22 expression
MAPKs are important intracellular signaling networks that eukaryotic cells use to transduce signals which are triggered by a wide range of extracellular stimuli. It is reported that MAPKs (p38, ERK, and JNK) play a crucial role in the induction of some ISGs (Katsoulidis and others 2005; Li and others 2010; Park and others 2011). To examine the role of these MAPKs in IFN-γ-induced TRIM22 expression, pharmacological inhibitors SB203580 (a p38 MAPK inhibitor), U0126 (an ERK1/2 inhibitor), and SP600125 (a JNK inhibitor) were utilized. As shown in Fig. 4A, IFN-γ induced the obvious phosphorylation of p38, ERK, and JNK in HepG2 cells, and the activation of these MAPKs could be efficiently suppressed by their corresponding inhibitors, as expected. However, our results showed that treatment of HepG2 cells with SB203580, U0126, or SP600125 failed to inhibit IFN-γ-induced TRIM22 expression at both mRNA and protein levels (Fig. 4B, C), indicating that the activity of p38, ERK, and JNK MAPK was not required for IFN-γ-induced TRIM22 expression.
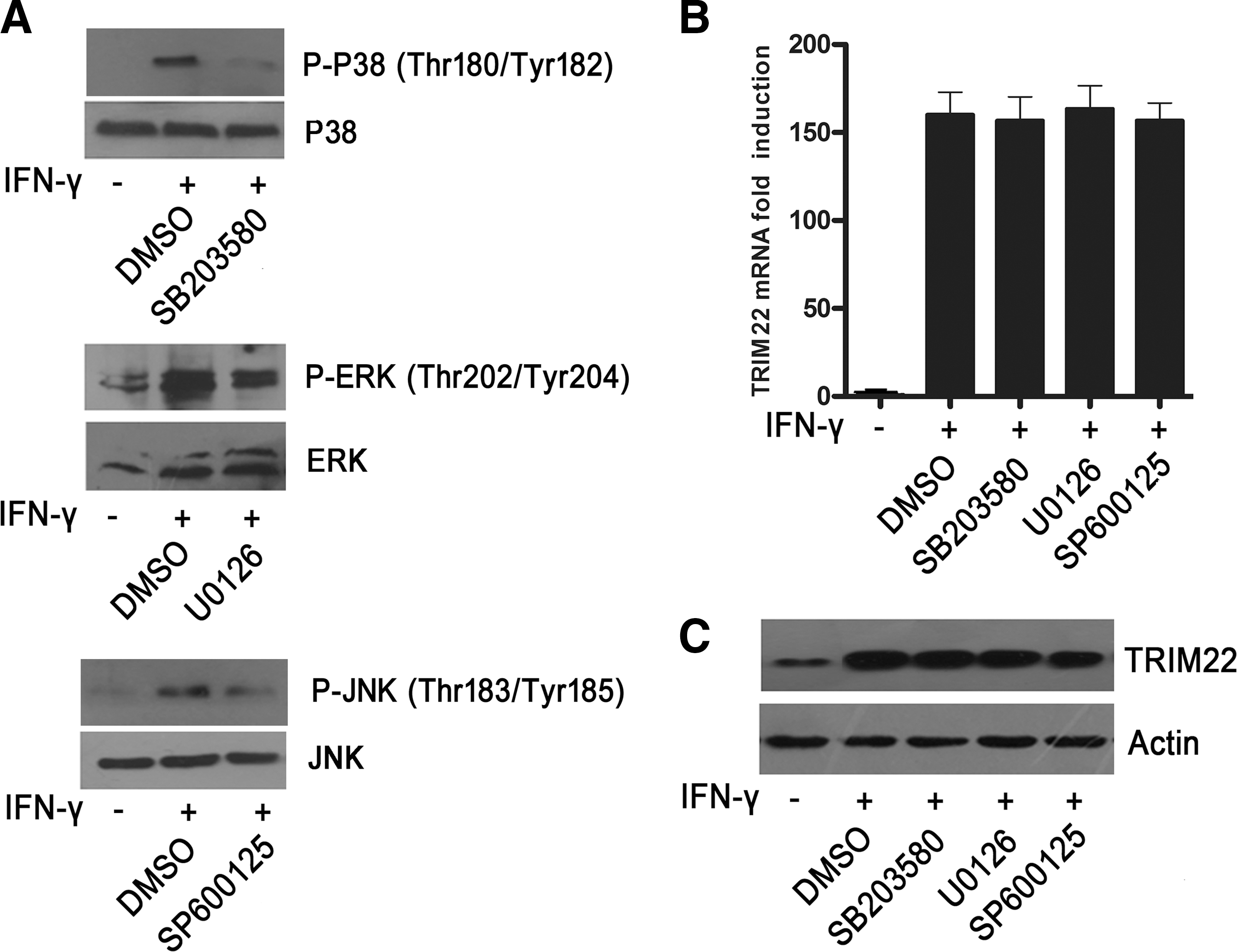
IFN-γ induces the activation of MAPKs in HepG2 cells, and lack of inhibition by SB 203580, U0126, and SP600125 on IFN-γ-induced TRIM22 expression.
IFN-γ induces the activation of pI3K/Akt/mTOR signaling pathway in HepG2 cells, and lack of inhibition by Ly294002, Akt inhibitor, and rapamycin on IFN-γ-induced TRIM22 expression
pI3K/Akt/mTOR signaling pathway has recently been demonstrated to be important for the induction of various genes in response to cytokine stimulation (Kaur and others 2005; Fang and others 2006; Venkatesan and others 2006; Kaur and others 2007). To evaluate the role of pI3K/Akt/mTOR signaling pathways in TRIM22 induction by IFN-γ, pI3K inhibitor (Ly294002), Akt inhibitor (1L-6-Hydroxymethyl-chiro-inositol2-[(R)-2-O-methyl-3-O-octadecylcarbonate]), and mTOR inhibitor (rapamycin) were used. We first determined the effect of IFN-γ on the activation of AKT and the mTOR, and the efficacy of their corresponding inhibitors. We observed that IFN-γ stimulation significantly increased the phosphorylation of AKT and the mTOR substrate p70s6k, while it had no effect on their expression levels (Fig. 5A, B). Expectedly, the IFN-γ-induced phosphorylation of AKT could be blocked by ly294002 and Akt inhibitor, respectively (Fig. 5A), and that of p70s6k could be markedly suppressed by ly294002, Akt inhibitor, and rapamycin, respectively (Fig. 5B). However, further investigations revealed that none of these inhibitors suppressed IFN-γ-induced TRIM22 expression at both the mRNA and protein levels (Fig. 5C, D).

IFN-γ induces the activation of pI3K/Akt/mTOR pathway in HepG2 cells, and lack of inhibition by Ly294002, Akt inhibitor, and rapamycin on IFN-γ-induced TRIM22 expression.
Inhibition of JAK, PC-PLC, or PKC activity suppresses IFN-γ-induced TRIM22 expression at the transcriptional level
We further investigated the effect of the pharmacological inhibitors mentioned earlier on IFN-γ-induced TRIM22 transcriptional activity by transfecting a TRIM22 promoter-dependent luciferase plasmid (pLUC160) into HepG2 cells. At 24 h posttransfection, cells were treated with various inhibitors for 30 min, followed by IFN-γ treatment for another 24 h. Consistent with their effect on IFN-γ-induced TRIM22 mRNA and protein expression, tyrosine kinase inhibitors genestein, tryphostin AG126, JAK inhibitor I, PC-PLC inhibitor D609, and PKC inhibitors staurosporine, Ro 31-8220 significantly inhibited IFN-γ-induced transcriptional activity of the TRIM22 gene, whereas other indicated inhibitors had no significant effect (Fig. 6A).
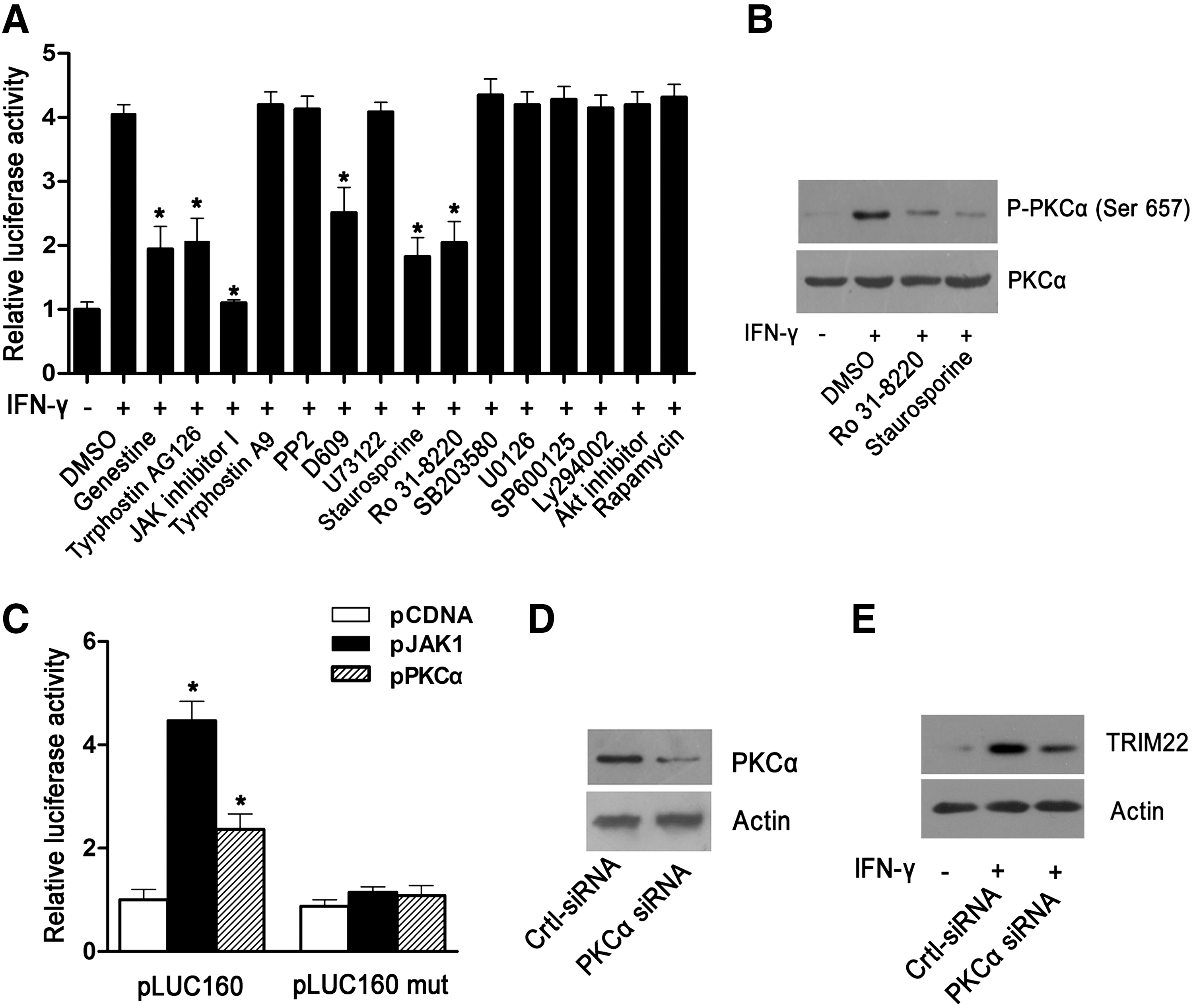
Inhibition of JAK, PC-PLC, or PKC activity inhibits IFN-γ-induced TRIM22 expression at the transcriptional level.
More than 12 PKC isozymes, the conventional PKC (PKCα, β1, β2, and γ), novel PKCs (PKCδ, ɛ, η, θ, and μ), and atypical PKCs (PKCζ and τ/λ) have been found (Newton 2001), of which PKCα/β2 (Nikodemova and others 2007) and PKCδ (Deb and others 2003) are known to be activated by IFNγ stimulation. Several studies have reported that PKCα was strongly expressed in HepG2 cells, whereas other PKC isozymes were expressed at relatively low levels or were not detected (Ducher and others 1995; Osaki and others 2004). We, thus, focus our attention on the role of PKCα in IFN-γ-induced TRIM22 expression in the present investigation. We first investigated the activation of PKCα in IFN-γ-treated cells, and also in cells pretreated with staurosporine or RO 31-8220. Results showed that IFN-γ significantly increased the phosphorylation of PKCα in HepG2 cells, which could be efficiently suppressed by staurosporine or Ro 31-8220, as expected (Fig. 6B). Next, we transfected the TRIM22 promoter plasmid pLUC160 into HepG2 cells along with plasmids encoding JAK1 or PKCα. Results showed that overexpression of JAK1 or PKCα significantly stimulated TRIM22 promoter activity (Fig. 6C). Our previous study showed that the cis-element, 5′eISRE, was crucial for TRIM22 transcription; we, thus, further investigated the role of 5′eISRE in JAK1-, and PKCα-mediated TRIM22 promoter activation by using a 5′eISRE-mutated TRIM22 luciferase reporter plasmid, pLUC160 mut (23). Results showed that, once the 5′eISRE was mutated, both JAK1 and PKCα failed to stimulate TRIM22 promoter activity (Fig. 6C). To further confirm the role of PKCα in IFN-γ-induced TRIM22 expression, we knocked down the expression of PKCα using siRNA. As shown in Fig. 6D, the PKCα-specific siRNA markedly down-regulated PKCα protein expression in HepG2 cells. Of importance, consistent with the effect of pharmacological inhibitors (stausporine and RO 31-8220), the knockdown of PKCα by siRNA significantly down-regulated IFN-γ-induced TRIM22 protein expression (Fig. 6E).
Inhibition of JAK, PC-PLC, or PKC activity attenuates IFN-γ-induced IRF-1 expression
Our previous data revealed that IRF-1 played a crucial role in IFNs-induced TRIM22 transcriptional activity via binding to 5′eISRE (Gao and others 2010; Wang and others 2011). Figure 7A also showed that inhibition of IRF-1 using siRNA strongly decreased IFN-γ-induced TRIM22 protein level. It is, therefore, of interest to know the role of JAK and PC-PLC/PKC pathways in IFN-γ-induced IRF-1 expression. The binding of STAT1 to a GAS element on IRF-1 promoter is reported to be indispensable for IFN-γ-induced IRF-1 expression (Pine and others 1994; van Boxel-Dezaire and Stark 2007). Thus, we first investigated the effect of JAK inhibitor I, PC-PLC inhibitor D609, and PKC inhibitors (staurosporine, Ro 31-8220) on the recruitment of STAT1 to IRF-1 promoter region by ChIP analysis. Results showed that treatment of HepG2 cells with JAK inhibitor I led to a nearly completed inhibition of STAT1 recruitment to IRF-1 promoter, and D609, staurosporine, Ro 31-8220 also significantly inhibited this event (Fig. 7B). Next, we investigated the effect of the inhibition of JAK and PC-PLC/PKC activity on IRF-1 induction. Consistent with the ChIP results, JAK inhibitor I abrogated IFN-γ-induced IRF-1 expression at both mRNA and protein levels, and D609, staurosporine, Ro 31-8220 also significantly suppressed IRF-1 induction (Fig. 7C, D). It should be noted that these pharmacological agent-mediated inhibitory effects on IFN-γ-induced IRF-1 expression were consistent with those on TRIM22 induction. To further confirm the role of PC-PLC/PKC pathway in IFN-γ induction of IRF-1, we examined the effect of inhibition of PKCα using siRNA on IRF-1 expression. As shown in Fig. 7E, the knockdown of PKCα significantly down-regulated the IFN-γ-induced IRF-1 expression.

Inhibition of JAK or PC-PLC or PKC activity attenuates IFN-γ-induced IRF-1 expression.
Discussion
Many TRIM members are regulated in response to IFN stimulation, supporting their role in antiviral and immunomodulatory activity (Carthagena and others 2009). Others and we have previously reported that TRIM22 was one of the most strongly induced TRIMs by IFNs and was implicated in IFNs-mediated anti-viral activity. Recently, we have identified a novel cis-element named 5′eISRE, which plays a pivotal role in IFN-γ-induced TRIM22 expression via binding to IRF-1 in a BRG1-dependent fashion (Gao and others 2010; Wang and others 2011). In this study, we extended previous works and identified the signaling pathways involved in the IFN-γ induction of TRIM22.
With use of tyrosine kinase inhibitors genistein and tyrphostin AG126, we found that the activity of tyrosine kinase was required for TRIM22 induction by IFN-γ. Recent evidence indicates that in addition to JAK, tyrosine kinase SRC or PYK2 may also play important roles in IFNs signaling. JAK is required for IFN-γ to activate both STAT1 and STAT3 by tyrosine phosphorylation, while SRC family kinases are reported to activate STAT3, not STAT1 (Qing and Stark 2004). PYK2 activation by IFN-γ is reported to activate MAPKs, such as ERK2, and it may also amplify SRC-dependent STAT3 activation (Takaoka and others 1999; van Boxel-Dezaire and Stark 2007). Using JAK inhibitor I (a pan-JAK inhibitor), PYK2 inhibitor tyrphostin A9, and SRC inhibitor PP2, we found that tyrosine kinase JAK was essential for IFN-γ-stimulated TRIM22 expression; while PYK2 and SRC appeared to be dispensable for this event. Since our previous data also demonstrated that siRNA-mediated inhibition of STAT1, not STAT3, significantly attenuated IFN-γ-induced transcriptional activity of TRIM22 (Fig. S1), TRIM22 induction by IFN-γ was most likely through the activation of STAT1, not STAT3.
It is now widely believed that, in addition to JAK-STAT pathway, IFN-γ requires the cooperation of multiple distinct signaling pathways for a full IFN response. Evidence indicates that phospholipases may play an important role in IFN signaling pathways (Sands and others 1994; Chang and others 2002; van Boxel-Dezaire and Stark 2007; Tsai and others 2009). Phospholipases function in modulating cellular responses to extracellular stimuli by generating various intracellular lipid signaling molecules through hydrolysis of membrane phospholipids. PI-PLC hydrolyzes 4,5-bisphosphate (PIP2) and generates DAG directly. DAG can either be generated along with phosphocholine through hydrolysis of choline-containing phospholipids by PC-PLC or indirectly formed by reactions involving PC-PLD and phosphatide phosphohydrolase (PAP). Usually, DAG is produced by the action of PI-PLC. However, PC-PLC may also be involved in the production of DAG, and it may contribute to the induction of some ISGs (Nishizuka 1995; Brindley and others 2009). In this investigation, we found that PC-PLC inhibitor D609 significantly inhibited IFN-γ-induced TRIM22 expression, while PI-PLC inhibitor U73122 and PAP inhibitor propranolol had no such effect. Since PLC-derived DAG is a well-established activator of PKC (Brindley and others 2009), we, thus, further investigated the role of PKC in TRIM22 induction using PKC inhibitors staurosporine and Ro 31-8220. It was found that the activation of PKC was required for IFN-γ induction of TRIM22. A further analysis showed that the knockdown of PKCα using siRNA also significantly inhibited IFN-γ-induced TRIM22 expression. Together, these data indicated that, in addition to JAK/STAT1, the PC-PLC/PKC pathway might also be involved in IFN-γ induction of TRIM22 in HepG2 cells. MAPKs and pI3K/AKT/mTOR signaling pathways have recently been revealed to be important for the induction of many ISGs. In the present investigation, we observed that IFN-γ stimulated the activation of these kinases in HepG2 cells; however, our data did not support their involvement in TRIM22 induction by IFN-γ.
We further investigated the effect of various phamarcological inhibitors on IFN-γ-induced TRIM22 promoter activity. Consistent with their role in IFN-γ-induced TRIM22 protein and mRNA expression, inhibitors of JAK and PC-PLC/PKC pathways showed a significant suppressive effect on IFN-induced TRIM22 promoter activity, and we further demonstrated the crucial role of 5′eISRE in JAK1- or PKCα-mediated TRIM22 activation. Given the importance of the association of 5′eISRE with IRF-1 in TRIM22 transcriptional induction, we further investigated the role of JAK and PC-PLC/PKC pathways in IFN-γ-induced IRF-1 expression. We found that, in addition to JAK, the PC-PLC/PKC pathway was also involved in IFN-γ-induced IRF-1 expression as demonstrated by the inhibitory effect of D609, staurosporine, and Ro 31-8220 on STAT1 recruitment to IRF-1 promoter and IRF-1 expression levels. Furthermore, the inhibition of PKCα by siRNA was found to significantly down-regulate IFN-γ-induced IRF-1 protein in the present investigation. There are conflicting reports regarding the role of PKC activity in IFN-γ-induced IRF-1 expression. There are reports that the inhibition of PKC activity leads to significant down-regulation of IRF-1 mRNA and protein expression in response to IFN-γ stimulation (Nikodemova and others 2007; Jeong and others 2009), and it is also suggested that PKC may affect IRF-1-dependent gene transcription through posttranslational regulation of IRF-1, such as phosphorylation and acetylation (Giroux and others 2003). We cannot exclude the possibility that PKC may have an effect on IRF-1 posttranslational modification, such as phosphorylation, in HepG2 cells. However, our current data indicated that the down-regulated IRF-1 expression level may be the main reason for the inhibitory effect of PKC inhibitors on TRIM22 expression, as the extent of PKC inhibitors (staurosporine and Ro 31-8220)-mediated inhibitory effect on IFN-γ-induced TRIM22 expression was closely correlated with that on IRF-1 induction.
In summary, our data indicated that in addition to the classical JAK pathway, the PC-PLC (not PI-PLC)/PKC pathway also played an important role in IFN-γ induction of TRIM22, which involved the cis-element 5′eIRSE and transcriptional activator IRF-1. Although MAPKs (p38, ERK, and JNK) and pI3K/Akt/mTOR pathways were efficiently activated in response to IFN-γ stimulation in HepG2 cells, these signaling pathways appeared to be not involved in IFN-γ-induced TRIM22 expression. These data may help further elucidate the molecular mechanisms of TRIM22 induction by IFNs and provide useful clues for TRIM22 induction by other stimuli such as viral infetion, LPS, or p53 protein, and might also be useful for designing a TRIM22-based antiviral strategy.
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81072428, 30890141), Major State Basic Research Development Program of China (2013CB530501), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT-IRT1075), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu “Pan-Deng” Project (BK2010004), the Natural Science Foundation of the Jiangsu Higher Education Institutions (11KJA180003), the Qing Lan Project of the Jiangsu Higher Education Institutions, and the Shanghai STC grant 09JC1401800.
Author Disclosure Statement
The authors have no conflicting financial interests.