
Abstract
Macrophages play an essential role in the innate immune response to infection and tissue injury. However, excessive macrophage activation may also significantly contribute to chronic inflammatory diseases. The Toll-like receptor (TLR) family are key regulators of innate immune responses in macrophages, and they are able to promote their survival and resistance against apoptosis. We, and others, have shown that the adaptive response gene, activating transcription factor 3 (ATF3), acts as a negative regulator of TLR signaling by repressing transcription of pro-inflammatory cytokines in primary mouse macrophages. Here, we describe a novel role for ATF3 as a component of TLR-mediated survival in macrophages. ATF3-deficient bone marrow macrophages show reduced survival in response to a range of TLR ligands and significantly higher apoptotic rates were observed in response to lipopolysaccharide, indicating that ATF3 is required to suppress apoptosis in macrophages. Furthermore, we show that ATF3 lies downstream of JNK signaling after TLR engagement, resulting in repression of pro-apoptotic Bak and Bax transcription.
Introduction
M
Toll-like receptors (TLR) are an evolutionarily conserved family of receptors that are capable of sensing molecular patterns associated with pathogens, and they act as important regulators of macrophage function. TLRs have been extensively characterized for their ability to signal pro-inflammatory cytokine production in a wide range of cell types and macrophages in particular. The majority of TLRs recruit the MyD88-IRAK-TRAF6 pathway, culminating in the activation of nuclear factor-κB (NF-κB), which drives the transcription of pro-inflammatory genes, including interleukin (IL)-6, IL-12, and tumor necrosis factor α (TNF-α). TRAF6 acts as a point of divergence in this pathway, as it can also recruit and activate p38, ERK, and JNK MAPK pathways, commonly resulting in the activation of AP-1-mediated transcription. Earlier, we, and others, found that activating transcription factor 3 (ATF3) was induced by TLR signaling in primary mouse macrophages and human dendritic cells (DC) (Gilchrist and others 2006; Whitmore and others 2007). ATF3 was able to inhibit transcription of IL-6, IL-12b, and IL-12p40, identifying it as a key regulatory factor limiting excessive activation of macrophages and DCs following recognition of pathogenic constituents (Gilchrist and others 2006; Whitmore and others 2007). However, the mechanism through which ATF3 is induced by TLRs has not yet been characterized. Evidence points to roles for NF-κB and AP-1 in ATF3 induction after TLR signaling, as consensus sequences for these transcription factors exist in the ATF3 promoter; while ATF2 and c-Jun were shown to directly activate the ATF3 promoter (Liang and others 1996). In addition, the MAPK pathways, which lie upstream of AP-1, are also likely to play a role in ATF3 induction after TLR signaling, as p38, ERK, and JNK have each been implicated in ATF3 induction in varying contexts (Liang and others 1996; Cai and others 2000; Keeton and others 2005; Lu and others 2007).
Apart from their role in the induction of pro-inflammatory cytokines, TLRs are also known to trigger pro- or anti-apoptotic responses in a cell-type-dependent manner. Bacterially derived lipopolysaccharide (LPS), which is recognized by TLR4, induces an apoptotic response in endothelial cells (Choi and others 1998); whereas in monocytes, neutrophils, or macrophages, LPS is known to protect against apoptosis (Goyal and others 2002; Ward and others 2005; Lombardo and others 2007). MyD88 is a critical component of TLR signaling, and deletion of this gene in bone marrow-derived macrophages (BMMs) results in complete abrogation of TLR4-mediated survival (Lombardo and others 2007). In addition, survival signaling mediated by TLRs depends on ERK and PI3K activation (Sester and others 1999; Monick and others 2000; Lombardo and others 2007). NF-κB is also an important mediator of TLR-mediated survival (Pagliari and others 2000; Lombardo and others 2007). In any case, it is likely that TLR signaling serves to counteract apoptosis after withdrawal of colony stimulating factor 1 (CSF-1) in the periphery (Lombardo and others 2007), an important growth factor critical for the maintenance of macrophage proliferation, survival, and differentiation (Pixley and Stanley 2004). CSF-1 starvation is characterized by caspase activation and cytochrome c release from the mitochondria, corresponding to a significant decrease in Bcl-XL expression (Hundal and others 2003), whereas TLRs have been demonstrated to up-regulate anti-apoptotic mediators such as cIAP2 (Cui and others 2000; Conte and others 2006), Bcl-XL (Okada and others 1998), and A1 (Pagliari and others 2000). Furthermore, survival mediated by LPS appears not to be associated with an increase in proliferation, as LPS does not induce the mitotic marker PCNA (Lombardo and others 2007).
Evidence suggests that ATF3 plays varied roles in cell death and cell-cycle progression; these are important factors in the context of cancer development in which these studies were conducted. For example, ATF3 over-expression in human PC3 prostate cancer cells has been shown to result in increased apoptosis (Huang and others 2008), while over-expression of ATF3 was shown to promote G1-to-S growth phase transition and proliferation in Du-145 human prostate cancer cells (Pelzer and others 2006). Clearly, uncertainty still remains as to whether ATF3 is either pro-apoptotic or anti-apoptotic, and this likely depends on the cellular context in question. In this regard, a few studies have identified ATF3 target genes that may regulate these processes. Since ATF3 is induced by TLR signaling, we were interested in determining whether ATF3 could regulate the survival of macrophages and the transcriptional targets that were responsible for this process.
The balance between apoptosis and survival is primarily controlled by members of the Bcl family of proteins, which localize to the mitochondrial outer membrane and act to control mitochondrial integrity and release cytochrome c, leading to activation of caspases, which enact apoptotic-specific proteolysis and cell death (Burlacu 2003; Opferman and Korsmeyer 2003). The Bcl family includes both pro-apoptotic and anti-apoptotic members (Opferman and Korsmeyer 2003). Of these, the pro-apoptotic members Bak and Bax are critical components of the mitochondria-dependent apoptosis pathway, the absence of which results in profound resistance to a wide range of apoptotic stimuli (Wei and others 2001). A recent microarray analysis has revealed that Bak and Bax are repressed by LPS stimulation, suggesting that these factors are responsible for modulating macrophage survival after TLR signaling. However, it is not known how TLRs are able to influence Bak and Bax transcription. Here, we provide evidence that ATF3 is specifically induced by TLRs downstream of JNK, resulting in macrophage survival and inhibition of apoptosis through the suppression of Bak and Bax.
Materials and Methods
Cell lines and reagents
RAW264.7 cells (ATCC) were grown in RPMI 1640+L-glutamine medium (Invitrogen) complemented with 1× antibiotic/antimycotic solution (Invitrogen) and 10% sterile FBS (ICPBio) at 37°C in a humidified atmosphere containing 5% CO2. The following TLR agonists were used at the concentrations indicated: LPS from Escherichia coli Serotype O111:B4 (TLR4 agonist; Enzo Life Sciences); poly I:C (pIC, TLR3 agonist; Roche); GpG ODN1826 (TLR9 agonist; Invivogen); Pam3CSK4 (TLR2 agonist; Invivogen); CL75 (TLR7 agonist; Invivogen); and Gardiquimod (TLR7 agonist; Invivogen). The following inhibitors were used dissolved in DMSO (Ajax Finechem) at the concentrations indicated: Actinomycin D (Sigma); SB203580 (p38 inhibitor; Calbiochem); JP600125 (JNK inhibitor; Calbiochem); and PD98059 (MEK inhibitor; Calbiochem). Cyclohexamide was purchased from Sigma.
Isolation of primary BMMs
Wild-type (WT) and ATF3 knockout (KO) mice were sacrificed by CO2 asphyxiation. Femur bones were resected, washed briefly with 70% ethanol, and then, sterile phosphate-buffered saline (PBS), before being placed in RPMI 1640+L-glutamine (Invitrogen) supplemented with 10% FBS (ICPBio) and 1× antibiotic/antimycotic solution (Invitrogen). Bones were cut at each end and flushed with a 23G needle to collect the marrow. Cells were homogenized with a 19G needle, centrifuged at 1,200 rpm for 3 min, and resuspended in fresh RPMI 1640+L-glutamine (Invitrogen) supplemented with 10% FBS (ICPBio), 1× antibiotic/antimycotic solution (Invitrogen) at 20% L-cell conditioned medium, as a source of CSF-1. Cells were plated in 10 cm nontreated polystyrene culture dishes (Sarstedt) for 5 days at 37°C in a humidified atmosphere containing 5% CO2, after which medium was replaced with fresh preparation as described earlier, and cells were maintained in culture for an additional 24 h. For experiments, cells were scraped, collected from plates, and resuspended in fresh RPMI, as earlier, but lacking L-cell conditioned medium. See next for the relevant seeding densities. For 96-well dishes, cells were cultured in 250 μL of medium, while in 6-well dishes, a volume of 2 mL was used. Cells were allowed to rest for 16 h before the addition of treatments. We reasoned that this would eliminate residual activation of CSF-1-activated pathways from our analysis, which is consistent with earlier publication (Lombardo and others 2007). This resembles the physiological conditions present, as macrophages traffic the periphery and encounter pathogenic organisms, enabling them to elicit sustained immune responses against infection, where they would otherwise become apoptotic to maintain a homeostatic turnover rate when survival factors are not present.
Western blot
About 3×106 BMM RAW264.7 cells or BMMs were treated as indicated in 6-well dishes. After treatment, cells were washed twice with ice-cold PBS, and whole-cell extracts were prepared in RIPA buffer (Tris 50 mM, NaCl 150 mM, SDS 0.1%, sodium deoxycholate 0.5%, and Triton X-100 1%) supplemented with PMSF and protease inhibitor cocktail (Sigma). Proteins were separated using either 10%–12% SDS-PAGE as required and transferred to Imobilon-P (Millipore) membranes before staining with the relevant antibodies: rabbit anti-ATF3 polyclonal antibody (No. SC-19; Santa Cruz); mouse anti-β-tubulin monoclonal antibody (No. 322600; Invitrogen); rabbit anti-PARP monoclonal antibody (No. 9532; Cell Signaling); rabbit anti-Bak polyclonal antibody (No. 3814; Cell Signaling); rabbit anti-Bax polyclonal antibody (No. 2772; Cell Signaling); mouse anti-Bcl-2 monoclonal antibody (No. SC-7382; Santa Cruz); rabbit anti-Bcl-XS/L polyclonal antibody (No. SC-634; Santa Cruz); or rabbit anti-Bim polyclonal antibody (No. 2819; Cell Signaling).
Isolation of RNA and quantitative reverse transcription–polymerase chain reaction
For quantitative reverse transcription–polymerase chain reaction (qRT-PCR), 3×106 RAW264.7 cells or BMMs were treated in 6-well dishes, then lysed and total RNA was isolated using Nucleospin RNA II columns (Macherey-Nagel) according to the manufacturer's instructions. RNA was reverse transcribed using Superscript III (Invitrogen), and cDNAs were used for PCR with SYBR GreenER reagents (Invitrogen) on the iCycler iQ real-time PCR detection system (Bio-Rad). The abundance of each mRNA was normalized against Gapdh expression. The primers are summarized in Table 1.
Immunofluorescence and analysis of mitochondrial membrane potential
About 2×105 RAW264.7 cells were treated as indicated in 96-well dishes. After treatment, cells were washed twice with ice cold PBS, then fixed in 4% paraformaldehyde in PBS. Cells were then washed and permeabilized with PBS+0.01% Triton X-100. Cells were blocked using CAS blocker (Invitrogen), then stained overnight with 1:200 rabbit anti-ATF3 polyclonal antibody (No. SC-19; Santa Cruz). ATF3 antibodies were detected with 1:200 Alexa Fluor 594 nm conjugated goat anti-rabbit IgG antibodies (No. A11012; Invitrogen). Counterstaining of nuclei was performed using Hoechst stain. Mitochondrial membrane potential was assessed in live RAW264.7 cells using Mitotracker Red, and counterstained with Hoechst. Fluorescent images of cells were obtained and analyzed using the Cellomics Arrayscan VTI instrument (Thermo Scientific). For colocalization studies, the Arrayscan algorithms identified the proportion of cells that exhibited ATF3 fluorescence that colocalized with the nuclear Hoechst dye, as compared with cytoplasmic localization of ATF3 fluorescence. Data are expressed as the nucleus:cytoplasm ratio of 10,000 cells. Similarly, Arrayscan algorithms were able to gate the proportion of cells presenting with an arbitrarily defined low threshold of mitotracker red fluorescence, indicative of significant loss of mitochondrial membrane potential.
Cell viability and caspase 8 activity
About 5×105 BMMs were cultured in 96-well dishes and treated as indicated. To assess cell viability, the Vialight plus (Lonza) and Cell Titer-Blue (Promega) were used according to the manufacturer's instructions. Similarly, caspase 8 activity was assessed using the Caspase Glo 8 kit (Promega), according to the manufacturer's instructions. Relative luminescence (Vialight and Caspase 8 Glo) and fluorescence (Cell Titer-Blue) were quantified using a plate reader.
Flow cytometry
For the analysis of cell cycle, 3×106 BMMs were cultured in 6-well dishes, treated, collected, pelleted, and washed in PBS, before overnight fixation in 70% ethanol in PBS, at 20°C. Cells were then pelleted, resuspended at a concentration of 1×106 cells/mL in propidium iodide (PI) solution containing 50 μg/mL PI (Sigma); 0.1 mg/mL RNase A (Invitrogen); and 0.05% Triton X-100. Cells were incubated for 30 min at 37°C. After washes, cells were resuspended in PBS and analyzed using the BD FACSAria flow cytometer. Unstained cells served as a control. Cell cycle was assessed by measuring PI fluorescence intensity peaks as an indicator of DNA content: 1× fluorescence intensity for G1/G0 cells; 2× fluorescence intensity for G2 phase cells; and between 1 and 2× intensity for S phase cells. Doublet cells were excluded following an assessment and adjustment of the pulse processing parameters. Apoptosis was assessed using an Annexin V apoptosis detection kit (No. 51-68981E; BD Pharmingen) according to the manufacturer's instructions. Cells were analyzed using the BD FACSAria flow cytometer and gated according to Annexin V-PE and 7-AAD-PI fluorescence intensity, with Annexin V+ cells being considered apoptotic.
Luciferase assay
HEK293T cells were transfected with a mixture of the Bak-luciferase reporter plasmid, pRL-TK renilla luciferase control plasmid, and either empty vector or Atf3 over-expression construct. The Bak-luciferase reporter construct consisted of a 1kb upstream sequence from the transcription start site cloned into a PGL-3 vector. The total amount of plasmid DNA transfected was equalized by the addition of empty control vector. After 24 or 36 h, cells were either left untreated or treated with LPS. Luciferase activity was measured with a dual-luciferase reporter assay system, according to the manufacturer's instructions (Promega) in a FLUOstar Optima plate reader (BMG Labtech). Data were normalized for nonspecific expression by the division of firefly luciferase activity with that of renilla luciferase.
Chromatin immunoprecipitation
Chromatin immunoprecipitation assays were performed according to the manufacturer's instructions (Millipore), with modifications. Cells (1×107) were treated with LPS at different time points, and then fixed with formaldehyde. After cell lysis, equal amounts of lysates were used for immunoprecipitation of chromatin with anti-ATF3, or immunoglobulin G. DNA was purified with a DNA purification column. The presence of the target gene promoter sequences in both the input DNA and the recovered DNA immunocomplexes were detected by quantitative PCR. PCR for Bak was performed using a SYBR Green PCR master mix (AP Biotech) on an iCycler iQ PCR machine (Bio-Rad). Primer pairs were: 5′-AGACCCTCACCTCCAAGGAT-3′ and 5′-CCTGCATCCTCCTTCTGTTC-3′ for the Bak promoter.
Results
ATF3 mediates macrophage survival
Our previous work, and that of others, has identified ATF3 as a key component of TLR signaling pathways. ATF3 is rapidly induced by different TLRs, including TLR4 (LPS) and TLR9 (CpG-DNA), and it functions as a negative regulator of NF-κB-driven transcription of target genes, including IL-6, IL-12p40, and TNF-α (Gilchrist and others 2006; Whitmore and others 2007). TLRs are known to be potent drivers of macrophage survival. In the absence of CSF-1 (the canonical macrophage growth and survival factor), LPS activation of TLR4 is able to induce macrophage survival (Lombardo and others 2007). ATF3 also has roles in apoptosis and cell-cycle regulation in varying contexts (Pelzer and others 2006; Huang and others 2008); however, this has not been demonstrated in macrophages. To determine whether ATF3 could play similar roles and drive alternate survival signaling pathways in macrophages, we generated BMMs from C57BL6/J mice (Materials and Methods section) and stimulated them with LPS or CSF-1 before quantification of Atf3 expression. As a control, we also measured cMyc after LPS or CSF-1 treatment. cMyc is a key cell-cycle regulator that is known to be induced by TLR4 to control macrophage survival, and a prominent target of CSF-1-mediated signaling. Accordingly, we observed potent induction of both Atf3 and cMyc by LPS (Fig. 1A), confirming previous reports by others (Myers and others 1995; Gilchrist and others 2006; Whitmore and others 2007). Interestingly, a potent increase in cMyc, but not Atf3, expression, was induced by CSF-1 (Fig. 1B), indicating that CSF-1 treatment was functional. However, this demonstrates that in the context of macrophage survival, ATF3 may be preferentially induced by LPS/TLR4 signaling, and not play a role in classical CSF-1-mediated survival.

Expression of Atf3 and cMyc after lipopolysaccharide (LPS) or colony stimulating factor 1 (CSF-1) treatment of bone marrow-derived macrophages (BMMs). BMMs were starved of CSF-1 for 16 h. Subsequently, BMMs were treated with either 100 ng/mL LPS
ATF3 protein is induced by LPS in a JNK-dependent pathway
To investigate the mechanisms of ATF3 induction after TLR4 activation, the RAW264.7 murine macrophage cell line was used to determine whether this occurred at the protein level, and whether this induction required de novo synthesis of Atf3 mRNA transcripts and protein, or stabilization of existing transcripts or protein. The results (Fig. 2A, B) show that ATF3 is induced at the protein level by LPS and that inhibition of transcription or translation, with actinomycin D and cyclohexamide, respectively, resulted in cessation of ATF3 induction. Thus, de novo synthesis of ATF3 occurs after TLR4 activation. In addition, after LPS stimulation, ATF3 readily translocated to the nucleus as demonstrated by immunofluorescence and analysis using an Arrayscan VTi high content instrument (Fig. 2C).
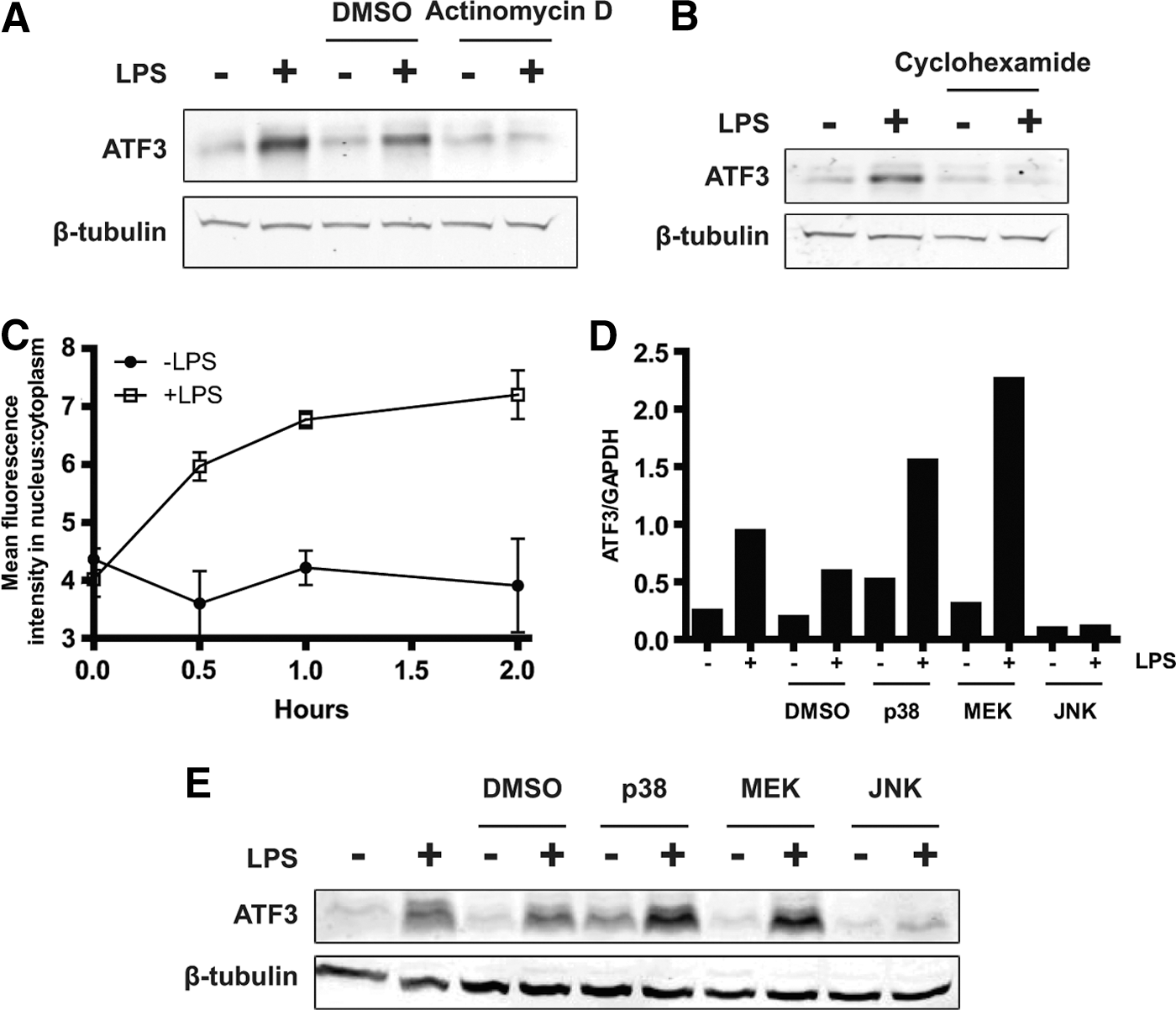
Activating transcription factor 3 (ATF3) synthesis is induced by LPS, downstream of JNK and translocates to the nucleus.
TLR activation causes a number of downstream signaling events, including the activation of MAPKs, which play roles in the survival of macrophages (Chang and Karin 2001; Parihar and others 2010). Interestingly, the MAPK pathways have been demonstrated to regulate the induction of ATF3 in response to a wide range of stimuli, in differing contexts (Cai and others 2000; Keeton and others 2005; Lu and others 2007). Therefore, to assess whether these pathways were involved in ATF3 induction after TLR4 activation, we pretreated RAW264.7 cells with small molecule inhibitors, including SB203580, PD98059, and SP600125, to disrupt p38, MEK, and JNK activity respectively, for 1 h before LPS stimulation. Only the JNK inhibitor was able to potently abrogate ATF3 induction at the transcript and protein level (Fig. 2D, E). Off-target effects were likely responsible for increased induction of ATF3 after p38 and MEK inhibition (Birkenkamp and others 2000). ATF3 induction is, therefore, likely to require the JNK pathway after TLR4 activation.
ATF3 contributes to macrophage survival following TLR stimulation
To assess the role of ATF3 in TLR4-mediated survival, we employed BMMs derived from ATF3-deficient (ATF3 KO) mice. Consistent with the lack of induction of Atf3 by CSF-1-mediated signaling and previous studies (Gilchrist and others 2006; Whitmore and others 2007), we were able to successfully differentiate ATF3-deficient bone marrow cells into macrophages using L-cell conditioned medium. WT and ATF3 KO BMMs were starved of L-cell supplemented medium for 16 h, then stimulated with LPS for 24 h. Both WT and ATF3 KO macrophages are less viable after 24 h if they are not stimulated, again illustrating that macrophages need a constant supply of survival signal to maintain their integrity over time (data not shown). However, WT macrophages retain viability after LPS stimulation, as measured by Vialight cell viability assay, confirming that LPS can promote survival of BMMs after CSF-1 withdrawal (Fig. 3). Importantly, ATF3 KO macrophages could not mount a survival response to LPS, with their level of viability matching the unstimulated controls. This suggests that ATF3 is required for TLR4-mediated survival of macrophages. WT and ATF3 KO macrophages were also stimulated with recombinant murine CSF-1 after the same period of L-cell supplemented medium withdrawal. These experiments demonstrate that ATF3 KO macrophages are competent in CSF-1-mediated survival signaling, though to a slightly lesser degree than their WT counterparts. This suggests that while ATF3 may play a small role in CSF-1-mediated survival signaling, this is in stark contrast to the effects seen in LPS-driven survival, where the ATF3 KO cells are completely unable to mount a response.

ATF3 is required for Toll-like receptor 4 (TLR4)-mediated survival of BMMs. Wild-type (WT) and KO BMMs were starved of CSF-1 for 16 h. After this, BMMs were incubated with or without 100 ng/mL LPS or 20% L-cell conditioned medium for 24 h. Cell viability was assessed using a Vialight assay. Triplicate values are presented as mean±SEM (***P<0.001, compared with corresponding WT value; unpaired t-test). Representative of 3 independent experiments.
Following our observation that ATF3 contributed to TLR4-mediated survival of macrophages, we were interested in assessing whether survival induced by other TLRs would also be affected, as ATF3 has been shown to also be induced by TLR3 and TLR9 (Whitmore and others 2007). Here, we show that in addition to LPS, TLR ligands, including Pam3Cys, pIC, CpG-DNA, and gardiquimod, are able to maintain viability of WT BMMs after L-cell supplemented medium withdrawal, as measured by Cell Titer-Blue viability assay (Fig. 4). In each case, ATF3 KO macrophages were deficient in mounting a survival response compared with WT cells. This suggests that ATF3 forms an important part of many TLR signaling pathways, which are each able to induce survival of macrophages in the absence of CSF-1 contained within L-cell supplemented medium.
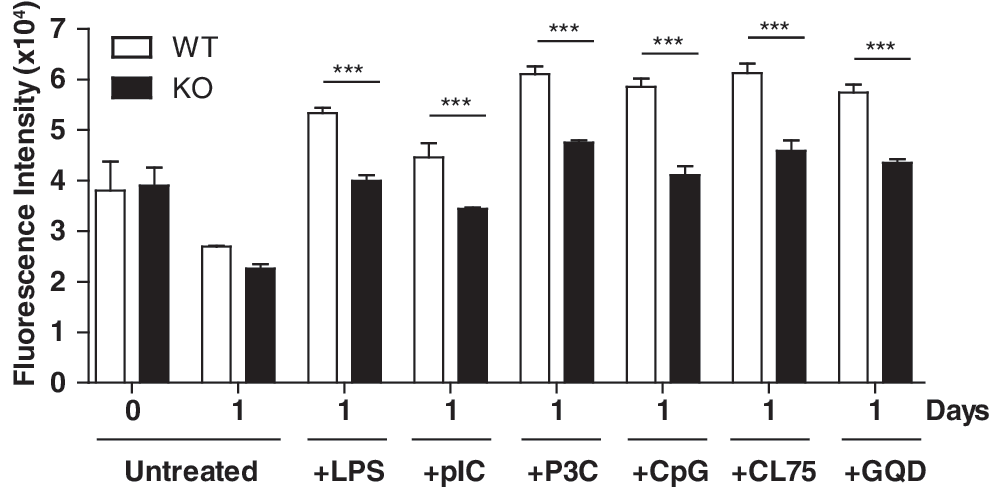
ATF3 is required for survival that is mediated by a range of TLRs. WT and KO BMMs were starved of CSF-1 for 16 h. Subsequently, cells were incubated with or without 100 ng/mL LPS, 100 ng/mL Pam3CysSK4 (P3C), 100 μM pIC, 1 μg/mL CL75, 10 μM CpG DNA (CpG), or 1 μM gardiquimod (GQD) for 24 h. Cell viability was analyzed using the Cell Titer-Blue viability assay. Triplicate values are presented as mean±SEM (***P<0.001, compared with their corresponding WT value; unpaired t-test). Representative of 3 independent experiments.
ATF3 suppresses apoptosis in stimulated macrophages
Given that our data demonstrate dependency on ATF3 to ensure viability of TLR-stimulated BMMs, we determined whether the decreased viability observed in ATF3 KO BMMs was the result of decreased cell-cycle transition or increased apoptosis. Accordingly, cellular DNA content was measured in PI-stained WT and ATF3 KO BMMs by flow cytometry to analyze cell-cycle status, after L-cell supplemented medium withdrawal and LPS treatment for 24 h. The results show (Fig. 5A) a significant proportion of WT and ATF3 KO cells in G1/G0 regardless of LPS treatment, indicating that a few were actively dividing. Compared with WT, the proportions of ATF3 KO G1/G0 and S phase cells remained relatively unchanged after LPS treatment. However, with G2 phase cells, a difference of approximately 6% (WT) versus 3.5% (ATF3 KO) demonstrated a relatively small contribution of ATF3 to cell-cycle progression. To investigate apoptosis, we treated WT and ATF3 KO BMMs with LPS before staining with annexin V antibody and 7-AAD dye. In response to LPS, WT BMMs showed a significant reduction in the proportions of apoptotic cells (annexin V positive, 7-AAD negative) (Fig. 5B, C). Conversely, ATF3 KO macrophages were not able to mount this response, with equal proportions of apoptotic cells present in both untreated and LPS-treated samples. This suggests that ATF3 is required to protect BMMs against apoptosis and to promote their survival in response to TLR4 activation. To complement this finding, lysates from WT and ATF3 KO BMMs were analyzed for the presence of 89 kDa PARP cleavage fragment, as a further indicator of apoptosis, after treatment with LPS for 24 h. The results (Fig. 5D) showed that WT cells had reduced PARP cleavage after LPS treatment, in contrast to ATF3 KO cells, which exhibited an increase in 89 kDa PARP fragment, which was indicative of higher amounts of apoptosis.

ATF3 protects against TLR4-induced apoptosis. WT and KO BMMs were starved of CSF-1 for 16 h and were then treated with or without 100 ng/mL LPS for 24 h.
Many apoptotic signals converge on the mitochondria, disrupting membrane potential and integrity in order to cease its energy production function and facilitate further downstream signaling, including the activation of apoptotic caspases. Accordingly, we analyzed the membrane potential of mitochondria in WT and ATF3 KO BMMs using Mitotracker Red. The proportions of cells with low Mitotracker Red fluorescence intensity, indicative of poor mitochondrial membrane potential, were significantly higher in ATF3 KO macrophages compared with WT (Fig. 5E). In accordance with this, the activity of a downstream effector of the apoptotic response, caspase 8, had increased activity (Fig. 5F). Overall, these results indicate that ATF3 is required for TLR-mediated survival, through its ability to suppress apoptosis.
ATF3 represses expression of the pro-apoptotic proteins Bak and Bax
The balance between apoptosis and survival is primarily controlled by members of the Bcl family of proteins that localize to the mitochondrial outer membrane and act to control mitochondrial integrity and release cytochrome c, leading to activation of caspases, which enact apoptotic-specific proteolysis and cell death (Burlacu 2003; Opferman and Korsmeyer 2003). Consequently, we investigated whether Bcl family members, both anti-apoptotic and pro-apoptotic, were differentially expressed in WT and ATF3 KO BMMs in response to LPS, and whether this correlated with the pro-apoptotic phenotype observed. Although pro-apoptotic Bak and Bax expression levels were reduced in WT BMMs 3 h after LPS stimulation, they were significantly up-regulated in ATF3 KO BMMs compared with WT in response to LPS (Fig. 6A, B). These data correlated with protein expression levels (Fig. 6C), whereby WT cells had a clear decrease in both Bak and Bax expression after 24 h of LPS treatment. Conversely, level of Bak and Bax protein were maintained after LPS treatment in ATF3 KO cells, suggesting that the increased level of apoptosis observed in these cells was the result of a dysregulated apoptotic response skewed toward cell death. The expression of anti-apoptotic proteins, Bcl-2 and Bcl-XL, and a pro-apoptotic BH3-only protein, Bim, remained unchanged in response to LPS in both genotypes. This suggests that ATF3 may be functionally important for the repression of Bak and Bax in response to TLR signaling. In order to test this, we transfected a Bak-luciferase reporter construct along with an ATF3 over-expression construct into TLR4-expressing HEK293T cells. Consistent with a direct role for ATF3 in Bak suppression, the over-expression of ATF3 resulted in significant repression of the Bak gene promoter (Fig. 6D). To determine whether ATF3 directly binds to the Bak promoter after LPS stimulation under physiological conditions, we also performed a ChIP assay, using anti-ATF3 antibody. Our results show that the Bak promoter only coprecipitates with specific antibodies against ATF3 after LPS stimulation (Fig. 6E, F). Binding occurred in the region amplified by primer set 2. Taken together, these results demonstrate that ATF3 is able to inhibit apoptosis in BMMs, after LPS stimulation, by repressing transcription of Bak and Bax. Furthermore, with regard to Bak, we have demonstrated that this repression is likely to be the result of direct binding of ATF3 to its promoter.
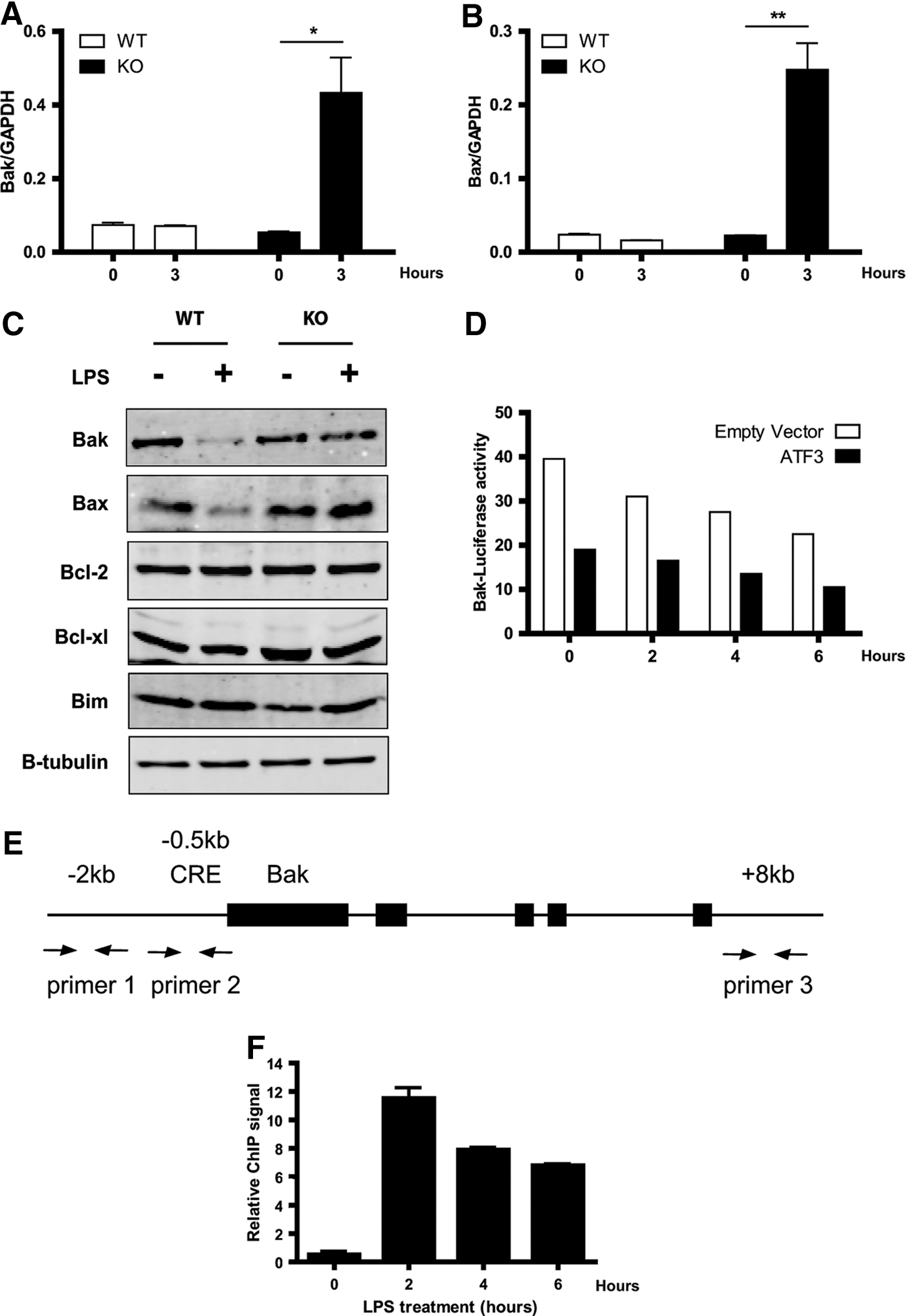
ATF3 represses Bak and Bax expression after TLR4 activation. WT and KO BMMs were deprived of CSF-1 for 16 h, then stimulated with or without 100 ng/mL LPS for 3 h. qRT-PCR was used to quantify Bak
Discussion
Macrophages represent one of the first lines of defense against invading pathogens, by rapidly responding to the threat and initiating the innate immune response. They also play varied roles in other physiological processes, including tissue repair, growth, and regeneration. The balance between apoptosis and survival is, therefore, critical for macrophage function, appropriate resolution of inflammation, and maintenance of tissue homeostasis. TLR signaling represents a key control component in macrophage function, which not only regulates immunomodulatory cytokine production, but also ultimately controls the fate of macrophages themselves, prolonging their responses appropriately in the presence of pathogen-associated molecular patterns or endogenous ligands that are indicative of tissue injury. In this study, we focused on determining the contribution of ATF3 to TLR-mediated survival of macrophages.
We showed that Atf3 is preferentially expressed in LPS-, but not CSF-1-, stimulated macrophages. Both TLR4 (Lombardo and others 2007) and CSF-1 (Pixley and Stanley 2004) signals are known to potentiate survival signaling in macrophages, and defects in CSF-1 signaling result in macrophage insufficiency. This is evident in the osteopetrotic op/op mouse, which lacks functional CSF-1 expression, resulting in significant deficiency of monocyte-derived macrophages (Felix and others 1990; Yoshida and others 1990; Naito and others 1991). Similarly, KO of the CSF-1 receptor results in a severe reduction in monocyte lineage cells (Dai and others 2002). Since Atf3 is not induced by CSF-1 and ATF3 KO are still able to mount survival responses, albeit to a lesser degree, after CSF-1 treatment, we conclude that ATF3 does not play a critical role downstream of CSF-1. Consequently, CSF-1 signaling remains largely intact in ATF3 KO macrophages. This is supported by the observation that ATF3 KO mice are phenotypically normal unless challenged (Hartman and others 2004).
ATF3 induction by TLR4 required de novo synthesis of ATF3, followed by nuclear translocation. Consistent with previous observations, components of the MAPK pathway were implicated in ATF3 induction. ATF3 has also been demonstrated to require the MAPK pathway for its induction in various other cellular contexts. For example, treatment of HUVEC and HAEC cells with homocysteine resulted in rapid induction of ATF3 that is mediated by the JNK MAPK pathway (Cai and others 2000). Furthermore, ATF3 induction in response to anisomycin, IL-1β, TNF-α, and H2O2, in HeLa cell cultures requires the p38 MAPK pathway (Lu and others 2007). Finally, induction of ATF3 by insulin, in rat H4IIE hepatoma cells, is dependent on signaling via the ERK family of MAPKs (Keeton and others 2005). Together, this evidence highlights that context dependency is paramount for the mechanism underlying ATF3 induction. Inhibition of the JNK pathway using the inhibitor SP600125 resulted in significant abrogation of ATF3 expression, demonstrating that JNK is critical for ATF3 induction by TLR4 (Fig. 2D, E). Since JNK has a defined role in macrophage survival (Himes and others 2006), this is consistent with ATF3 also regulating survival of macrophages. Whether JNK directly phosphorylates ATF3 has not been established, but tyrosine phosphorylation of ATF3 has been demonstrated downstream of IL-10 signaling (Stearns and others 2004), and ATF3 phosphorylation has also been detected in adenovirus-E1-transformed cells (Hagmeyer and others 1996). However, JNK and p38 MAPKs have been excluded as possible kinases for ATF3 phosphorylation in HEK293 transfection studies, even though they share a conserved JNK docking domain and phosphoacceptor site with JDP2 and c-Jun (Katz and Aronheim 2002).
We found that the activation of TLR4, −3, −9, −2/6, and −7 (by LPS, pIC, CpG-DNA, Pam3Cys, CL75, and gardiquimod, respectively) was able to promote the survival of WT BMMs in the absence of L-cell supplemented medium for 24 h. While similar responses have been observed after the activation of TLR4 (Lombardo and others 2007) and TLR9 (Sester and others 2006), we can now include TLR3, −2/6, and −7 as receptors that are able to promote the survival of macrophages after CSF-1 withdrawal. Importantly, we show that ATF3 KO BMMs possess a defect in their ability to mount survival responses after TLR4, −3, −9, −2/6 and −7 activation. This suggests that ATF3 is required for TLR-mediated survival in response to a wide range of ligands. It is likely that this effect would extend to other TLRs, including TLR5 and TLR8, as each of the TLRs share conserved signaling components. However, ATF3 only contributes in part to overall TLR-mediated survival, as deletion of ATF3 in BMMs resulted in partial inhibition of survival responses. NF-κB, ERK, and PI3K pathways are also implicated, as pharmacological inhibition of these before LPS treatment of BMMs has been shown to significantly reduce TLR-mediated survival (Lombardo and others 2007). Likewise, blockage of NF-κB, either pharmacologically or via IκBα super repressor over-expression, reduces the viability of RAW264.7 cells or human primary macrophages (Pagliari and others 2000). The pro-survival effects of NF-κB have largely been attributed to its transcriptional regulation of anti-apoptotic genes. These include Bcl-2, Bcl-XL, cIAP2, and Al (Chu and others 1997; Lee and others 1999; Tamatani and others 1999; Pagliari and others 2000). Consistently, cIAP2 (Cui and others 2000; Conte and others 2006), Bcl-XL (Okada and others 1998) and A1 (Pagliari and others 2000) have each been demonstrated to be induced by TLRs and may, therefore, be responsible, in part, for the pro-survival effect of TLR signaling in macrophages.
To determine the process through which ATF3 was able to promote TLR-mediated survival in BMMs, we analyzed cell-cycle progression as well as apoptosis in response to LPS. Proportions of G1/G0 and S-phase cells were not significantly different between WT and ATF3 KO BMMs after treatment with LPS. Although a significant difference in the proportions of ATF3 KO cells in the G2 phase compared with WT was observed, this is unlikely to contribute to the overall viability of these cells, considering the fact that G2-phase cells are still not actively dividing. The low proportions of actively dividing cells observed after treatment with LPS were consistent with a report demonstrating that LPS does not induce mitosis (Lombardo and others 2007). The role of ATF3 in cell-cycle control remains controversial. ATF3 over-expression has been demonstrated to promote DNA synthesis and cyclin D1 transcription in mouse hepatic tumor cells (Allan and others 2001). Conversely, over-expression of ATF3 in HeLa cells resulted in reduced cell growth and G1- to S-phase transition (Fan and others 2002). Similarly, ATF3 KO fibroblasts have been demonstrated to cycle from G2 to S phase more efficiently, implicating ATF3 as a negative regulator of cell-cycle progression; a process that is mediated by the suppression of cyclin D1 expression (Lu and others 2006). On the other hand, we showed that annexin V-positive apoptosis was significantly increased in ATF3 KO BMMs treated with LPS. This correlated with decreased mitochondrial integrity, and increased caspase 8 activity and PARP cleavage. In contrast, WT BMMs were able to actively down-regulate apoptosis after treatment with LPS, consistent with previous studies demonstrating the protective role of LPS in monocytes and macrophages (Goyal and others 2002; Ward and others 2005; Lombardo and others 2007). Taken together, these data implicate ATF3 as a negative regulator of apoptosis in BMMs after TLR signaling.
The Bcl-2 family of proteins constitutes key regulators of apoptosis. The tumor suppressor gene p53 has been shown to play an important role in the regulation of Bax expression, in its acetylated form (Tang and others 2008), and in the regulation of Bak expression (Graupner and others 2011). However, little is known about the expression of Bax or Bak in macrophages. Here, we demonstrated elevated transcriptional and concurrent protein expression of Bax and Bak in ATF3 KO macrophages. Down-regulation of the pro-apoptotic Bcl-2 family members, by ATF3 in macrophages, appeared to be specific for Bak and Bax, because we did not observe changes in an alternative apoptosis-promoting protein, Bim. Likewise, we did not observe any changes in Bcl-2 or Bcl-XL expression. Subsequent luciferase reporter and ChIP experiments support that ATF3 has a direct role in the transcriptional regulation of Bak. Together, these data suggest that Bak and Bax provide ATF3 with an essential gateway to regulate apoptosis and mediate TLR-induced survival. To date, ATF3 has also been demonstrated to regulate Bax expression in paclitaxel-treated HeLa cells, by binding to the promoter in combination with p73 (Oh and others 2008).
Overall, we have demonstrated an important role for ATF3 in the maintenance of macrophage survival in vitro, via its ability to inhibit apoptosis through its repression of Bak and Bax, likely at the transcriptional level. It is known that the deletion of ATF3 in murine macrophages results in exaggerated pro-inflammatory cytokine production after TLR stimulation, and in whole mouse experiments, it results in exquisite sensitivity to endotoxic shock and PR8 influenza infection (Gilchrist and others 2006; Whitmore and others 2007). The fact that our results show increased apoptosis of ATF3 KO macrophages in vitro seems counterintuitive, as an increase in macrophage apoptosis would logically limit the over-active inflammatory response in ATF3 KO mice. Therefore, it would be at first appropriate to assess whether macrophage apoptosis occurs to a greater degree after TLR stimulation in ATF3 KO mice in vivo. Evidently, ATF3 KO macrophages were shown to produce excessive inflammatory cytokines after LPS, pIC, or CpG-DNA after 20–24 h (Whitmore and others 2007); within the window of apoptosis induction observed in our experiments. Therefore, ATF3 KO macrophages may still be capable of enhancing inflammatory responses, irrespective of decreasing viability in vitro, suggesting that ATF3 KO macrophages will still be able to make significant contributions to over-active inflammation in vivo. Furthermore, it is not yet known as to what degree ATF3 may be influencing other components of the inflammatory response in vivo. Therefore, conditional deletion of ATF3 in macrophages would address this issue in whole animal models. Alternatively, the characteristics of in vitro generated BMMs are not likely to reflect the heterogenous and plastic populations of macrophages, including pro-inflammatory M1 and anti-inflammatory M2 polarized phenotypes in vivo. M1 macrophages can be elicited by LPS stimulation, while M2 macrophages are produced by IL-4 and IL-13 stimulation (Lawrence and Natoli 2011). Our experiments are likely to represent the contribution of ATF3 to apoptosis in M1 polarized macrophages only. Survival of ATF3 KO M2 polarized macrophages could, therefore, be assessed in vitro. Loss of ATF3 may potentially alter the balance of M1/M2 macrophages in vivo such that a predominant pro-inflammatory environment still exists, despite increased apoptosis of pro-inflammatory macrophages.
Footnotes
Acknowledgments
This work was supported by funding from the Australian National Health and Medical Research Council (1006588 to B.R.G.W. and D.X.) and the Victorian Government's Operational and Infrastructure Support Program. M.R.T. was supported by a Ron Evans Bowel Cancer Scholarship and an Australian Rotary Health Research Fund—District 9680 Bowelscan Scholarship.
Author Disclosure Statement
No competing financial interests exist.