
Abstract
Antibodies can be generated against a therapeutic protein upon administration to human subjects. When the therapeutic protein closely mimics one of the subject's endogenous proteins, those antibodies might bind to the endogenous protein in addition to the therapeutic protein. This scenario results when tolerance to the endogenous protein is broken. The consequences of breaking tolerance include an autoimmune response where antibodies are generated against the endogenous protein. These autoantibodies could have significant clinical relevance depending on several factors, including the redundancy of action of the endogenous protein as well as the concentration, binding affinity, and neutralizing potential of the antibodies. The consequences of a therapeutic-protein-induced autoimmune reaction can be challenging to manage as the stimulus for further perpetuation of the immune response can shift from the therapeutic protein to the endogenous protein. The potential for inducing an autoimmune response is one of the reasons that the immune response to a therapeutic protein should be monitored if it persists through the end of the study.
Introduction
A
It is widely recognized that therapeutic proteins administered to human subjects have the potential to elicit an immune response (Schellekens 2003). The nature of the immune response varies greatly depending on the therapeutic protein, its product quality attributes, dosing regimen, and the immune status of the subject. The effects of an immune response to a therapeutic protein range from no-observable clinical effect through complete neutralization of the therapeutic protein (Koren and others 2002). Most immune responses to therapeutic proteins are not classified as autoimmune responses because the anti-drug antibodies (ADAs) exclusively bind to the therapeutic protein. However, in cases where the therapeutic protein closely mimics an endogenous protein, the antibodies generated against the therapeutic protein might also bind and potentially neutralize the endogenous counterpart as well. This was demonstrated when thrombocytopenia occurred after subjects developed antibodies capable of binding a therapeutic protein (MGDF) as well as endogenous thrombopoietin (Li and others 2001). In these cases, the antibodies, while initially produced against the MGDF, were not able to distinguish that protein from its endogenous counterpart, thrombopoietin. The end result was that the subjects were no longer able to produce sufficient platelets because their endogenous thrombopoietin had been neutralized. When antibodies are produced that bind to an endogenous protein, it indicates that the normal state of tolerance to that endogenous protein has been broken which has allowed antibodies to be formed that can bind to “self-proteins.”
While it is not well understood why some therapeutic proteins have been able to break tolerance, recent reports suggest that high levels of aggregated therapeutic proteins may be a contributing factor (Kumar and others 2011). A recent article provides evidence that a possible mechanism for increased immunogenicity with aggregates is a longer residence time at the subcutaneous injection site and an increase in cytokine levels associated with subcutaneous injection of aggregated material (Filipe and others 2013). Another factor that has been linked to aggregates being associated with a greater rate of immunogenicity is that they may cause an increase in the inflammatory signal at the injection site that leads to an increase in recruitment of dendritic cells (Fathallah and others 2013). The possible role of aggregates is being widely investigated and the coming years will likely lead to a more clear understanding of any association between the presence of aggregates and the immunogenicity of therapeutic proteins.
There are other risk factors involved with initiating an immune response against a therapeutic protein that are also being studied; however, more work will be required before we can fully understand all the possible causes (Singh 2011). It is important to recognize that even when antibodies are formed that bind to endogenous proteins, not all cases result in a clinical effect. An immune response that receives T Cell help is much more likely to develop into a clinically meaningful autoimmune response. This T Cell help mediates immune maturation that can result in the production of a higher concentration of antibodies, an increase in the number of epitopes recognized by those antibodies, an increase in the binding affinity of the antibodies, and typically a shift in the class and subclass of antibodies that are produced. Without T Cell help, it is unlikely that an autoimmune response would rise to a level that could be observed clinically because it is less likely that an immune response that only includes IgM antibodies would have a significant clinical effect.
When Autoimmunity Could Result from an ADA Response
Autoimmunity is broadly defined as an immune response that is directed against self and, as such, any ADA response could be broadly interpreted as autoimmunity. It is important to recognize that there are several important factors that help us define when an ADA becomes a case of autoimmunity. The first 2 important factors to understand are if the therapeutic protein mimics an endogenous counterpart and if antibodies against the therapeutic can bind to that endogenous counterpart. Both must be the case for autoimmunity to be established. The risk of autoimmunity induction is greater when the therapeutic closely matches the endogenous counterpart. In cases where only a small portion of the therapeutic is identical to an endogenous protein, there is less chance that antibodies produced against that therapeutic would also bind to the endogenous protein, although epitope spreading could occur. Epitope spreading could result in a future generation of ADAs in the subject that would be able to bind to an epitope nearly identical or identical to one found on the endogenous counterpart.
The next fundamental question to be answered is can the ADA continue to be produced once the therapy is withdrawn? As was the case with the previously mentioned thrombocytopenia cases, these antibodies and resulting thrombocytopenia persisted after treatment was withdrawn. The implication is that endogenous thrombopoietin was able to continue to stimulate the immune system to continue to generate more antibodies even after the therapeutic protein was no longer available.
The ability to recognize when a therapeutic protein has induced autoimmunity depends upon the quality of the analytical procedures that are used to identify and characterize the ADAs. There are a wide variety of analytical procedures that are currently used (Thorpe and Swanson 2005). The availability of sensitive and reliable testing is critical. A comparison of ADA data with clinical effect is important to associate clinical consequences with the presence of an ADA response. The types of clinical response most often associated with ADAs that are part of an autoimmune response include loss of efficacy and/or rapid clearance of the therapeutic protein. Figure 1 demonstrates the effect of a clearing antibody that is able to rapidly clear the therapeutic protein from circulation. As shown in the figure, the serum concentration is much lower in the subject who is antibody positive. Another possible effect of an antibody is shown in Fig. 2 where the therapeutic protein is sustained longer in the circulation due to the presence of antibodies. In this example, the subject with antibodies has a higher circulating level of drug than the initial dose where the subject was negative for antibodies. The antibodies in this case are acting to prevent the drug from being cleared. Figure 3 shows the effect of a neutralizing antibody. The subject has a normal platelet count after the first 2 administrations of the therapeutic but the platelets are dramatically reduced by the third treatment and dropped to very low levels. The antibody in this case has neutralized not only the therapeutic protein but also endogenous thrombopoietin resulting in platelet deficiency (Li and others 2001). It is important to perform a neutralizing antibody test to understand whether the ADA can neutralize or block the biological effect of the therapeutic protein (Indelicato 2003). A neutralizing antibody is much more likely to induce autoimmunity because the epitope recognized by the ADA is in the biologically active region of the molecule that is likely conserved with the endogenous protein. Another factor that is important to monitor is the presence of any clinically relevant signal. In the case of antibodies against MGDF, the platelet count in the subjects dropped significantly as a result of the antibodies neutralizing the drug as well as endogenous thrombopoietin. It was this signal that confirmed the clinical relevance of the ADAs. Not all therapeutic proteins have such a clear clinical signal, but understanding the biological effect induced by the drug helps identify any clinical marker that should be monitored to assess the importance of an ADA response.
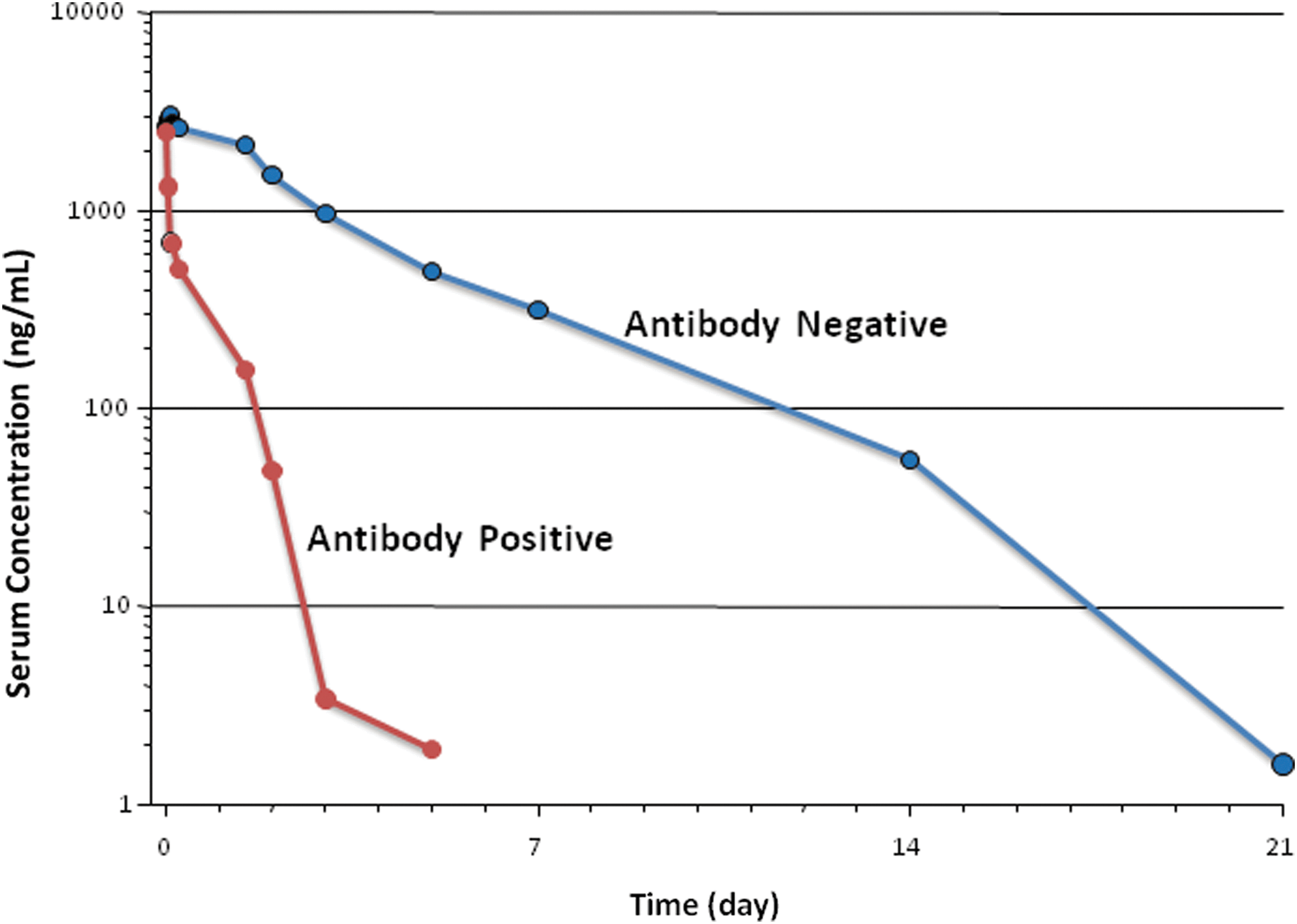
Shown is the pharmacokinetic effect of antibodies in a subject that develops antibodies capable of enhancing the clearance of the therapeutic protein. The blue line represents clearance prior to induction of an immune response. The red line shows the more rapid clearance of the drug in the subject when antitherapeutic protein antibodies are present.

Shown is the pharmacokinetic effect of antibodies in a subject that develops antibodies capable of inhibiting the clearance of the therapeutic protein and sustaining the therapeutic in circulation. The blue line represents clearance prior to induction of an immune response. The red line shows the decreased clearance of the drug in the subject when antitherapeutic protein antibodies are present.
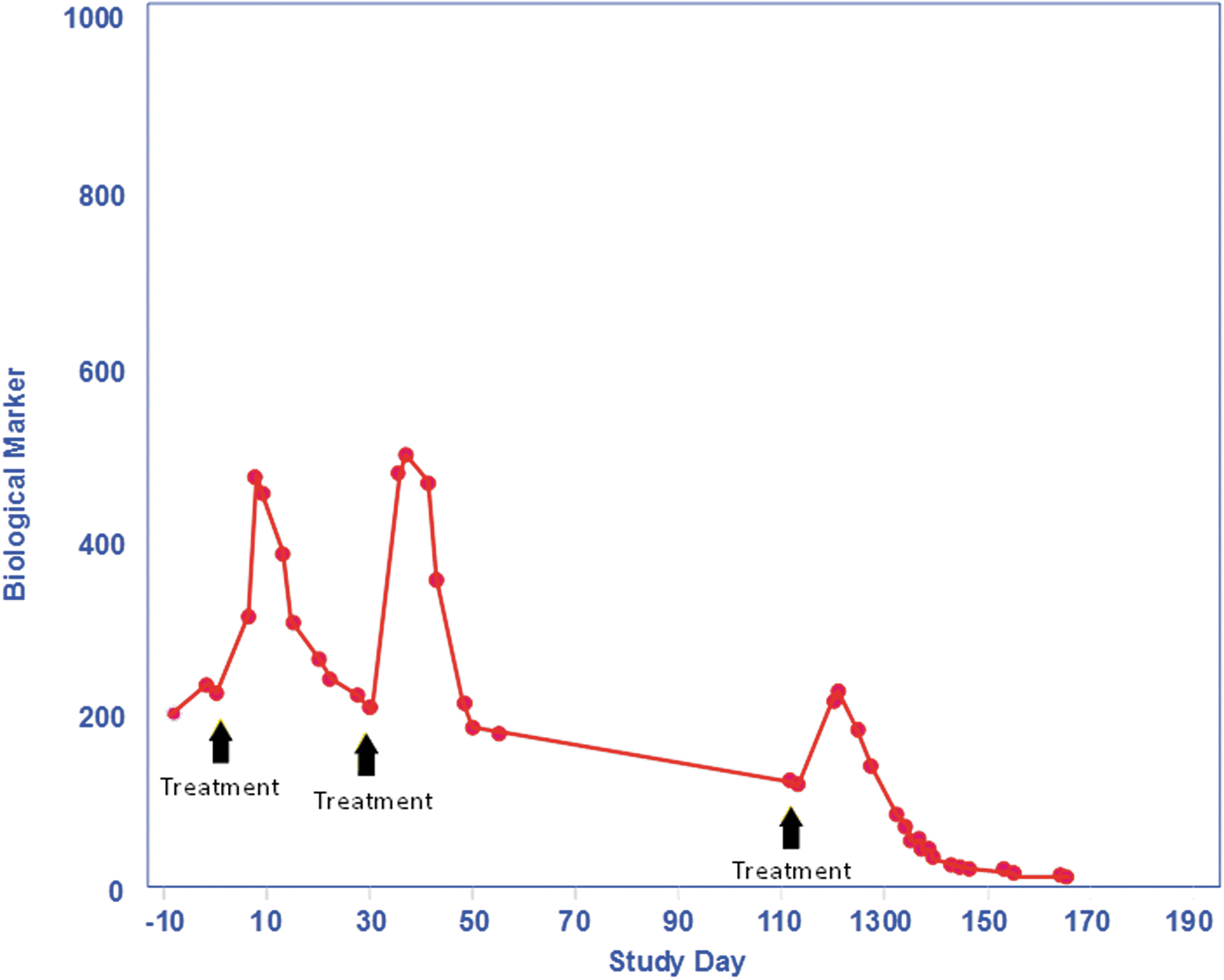
This figure shows the effect of a neutralizing antibody to thrombopoietin on platelet production. Antibodies against thrombopoietin are present following the second treatment and those antibodies abrogate the effect of the therapeutic protein to increase platelets at the third treatment. The antibodies also neutralize endogenous thrombopoietin as demonstrated by the rapid fall in platelets below treatment levels.
Therapeutic Proteins That Have Induced Autoimmunity
While most therapeutic proteins do not induce long-lasting immune responses that are classified as autoimmunity, there have been multiple examples. Additional therapeutics that have been associated with autoimmune diseases (in addition to MGDF that has previously been described) include erythropoietin (Casadevall and others 2002) and therapeutic enzymes that are used to treat lysosomal storage diseases, including Pompe's disease and Gaucher's disease (Wang and others 2008). What is common among these examples is the lack of redundancy in a biological system to produce the same effect as the therapeutic. Thrombopoietin is essential for platelet production just as erythropoietin is essential for red blood cell production. In cases where these are absent or their production is neutralized by an ADA, serious clinical consequences occur because there is no redundancy. Likewise with lysosomal storage disease, when these enzymes are not present or blocked, the consequences can be fatal.
Table 1 shows the characterization of anti-erythropoietin antibodies (Swanson and others 2004) from pure red cell aplasia subjects originally reported by Casadevall (Casadevall and others 2002). These antibodies range in concentration (relative to a rabbit polyclonal positive control antibody) from 4 to 43 mcg/mL. Of note, the isotypes of the anti-EPO antibodies were predominately of the IgG4 subclass. The presence of the large proportion of these antibodies being IgG4 suggests that these anti-EPO antibodies represent a well-established and mature immune response that would be consistent with an established autoimmunity case.
The characterization of anti-erythropoietin antibodies as performed by Biacore analysis obtained from subjects with pure red cell aplasia. The concentration of the antibodies is related to a concentration analysis of a rabbit polyclonal anti-erythropoietin antibody.
It is easiest to identify cases of therapeutic-protein-induced autoimmunity when the clinical signal is clear as described earlier. It is quite likely that there are other examples that have not been identified either due to biological redundancy that limits the clinical observations, or immunogenicity assessment that does not identify all ADAs. Other reported examples where an ADA response could be considered part of an autoimmune response include enzyme replacement therapy. Pompe disease is an example of a lysosomal storage disease that has been successfully treated with enzyme-replacement therapy. It was demonstrated that induction of immune tolerance to the replacement enzyme results in an enhanced response (Sun and others 2007). This provides additional evidence that the ability to reduce the immune response to a therapeutic protein that acts to supplement an endogenous protein could result in a better therapeutic outcome. Further evidence on the value of immune modulation regarding therapeutic-protein-induced autoimmunity was demonstrated by a combination therapy of rituximab plus methotrexate and intravenous gamma globulin used in an infantile Pompe's disease patient (Mendelsohn and others 2009). The result of the therapy was to reduce the antibody response to the replacement enzyme and provide clinical benefit from the replacement-enzyme therapy. This successful therapy suggests a potential path forward for other replacement therapeutic proteins in reducing or eliminating the consequences of autoimmunity induced after treatment with a therapeutic protein.
Future Steps
It is important to recognize that any therapeutic protein that mimics an endogenous protein has the potential to elicit an autoimmune response. This potential underscores the importance of fully understanding the immune response to therapeutic proteins by implementing a robust screening strategy and following up on all positive samples with a full characterization. Nonclinical studies, where immunogenicity rates are often quite high, can provide early evidence regarding the possible manifestations of an immune response in humans. This early evidence on potential serious consequences of an immune response, including loss of function of an endogenous counterpart, can allow early clinical studies to be designed with appropriate precautions including monitoring. It is important to recognize, however, that the presence of an immune response in a nonclinical study does not always correlate with a clinical immune response. Understanding the characteristics of the immune response, the magnitude, epitopes recognized, binding affinity, the timing of antibody production, as well as the class and subclass of the antibodies produced can be valuable in evaluating both how and why tolerance was broken. For example, it would be useful to know whether the original epitope recognized on the therapeutic protein was also present on the endogenous protein. This could help explain whether epitope spreading was involved in the breaking of tolerance or whether the initial recognition mediated the breakage. It would not be possible to answer that question unless samples were available and assays sufficiently sensitive were used to facilitate identifying the first antibodies produced against the therapeutic protein.
A careful comparison of antibody production against therapeutic proteins and the product quality attributes of the drug that the subject received could also provide insight into what event was responsible for the breaking of tolerance and initiation of an autoimmune response. It is only after we have a better understanding of why certain therapeutic proteins in certain subjects elicit autoantibodies that we will be able to manufacture therapeutic proteins that have a reduced risk of inducing autoantibodies. The important work in immune modulation with Pompe's disease patients underscores the value in tolerance induction as a potential strategy to reduce the impact of autoimmunity ADAs.
The field of immunogenicity assessment continues to improve. These improvements in analytical procedures will provide a better opportunity to identify those therapeutic proteins that induce autoimmunity, and possibly to eventually reduce both the immunogenicity and the consequences for patients.
Author Disclosure Statement
The author is a full time employee of Amgen, Inc.