
Abstract
Tumor necrosis factor α-related apoptosis-inducing ligand (TRAIL) is a potent inducer of apoptosis in Jurkat T lymphoma cells. One of the characteristics of the phase preceding overt apoptosis is the marked downregulation of protein synthesis. We have investigated factors that can influence this response and have explored some of the signaling pathways involved. We show that interferon-α (IFNα) pretreatment desensitizes Jurkat cells to TRAIL-induced inhibition of protein synthesis, such that the concentration of TRAIL required for 50% inhibition is increased by 10-fold. The inhibition of translation is characterized by dephosphorylation of the eIF4E-binding protein 4E-BP1 and IFNα desensitizes Jurkat cells to this effect. IFNα also inhibits TRAIL-mediated dephosphorylation of the growth-promoting protein kinase B (Akt). Since Jurkat cells are defective for phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and therefore have constitutive phosphoinositide 3-kinase (PI3K) activity, we investigated the consequences for protein synthesis of inhibiting PI3K using LY294002. Inhibition of PI3K partially inhibits translation, but also enhances the effect of a suboptimal concentration of TRAIL. However, LY294002 does not block the ability of IFNα to protect protein synthesis from TRAIL-induced inhibition. Data are presented suggesting that IFNα impairs the process of activation of caspase-8 within the TRAIL death-inducing signaling complex.
Introduction
P
Hyperphosphorylation of 4E-BP1 promotes the dissociation of this factor from eIF4E. Since 4E-BP1 binds to eIF4E in competition with the large scaffold protein eIF4G, dissociation of 4E-BP1 allows eIF4E to interact with eIF4G (Avdulov and others 2004; Clemens 2004). Together with eIF4A, eIF4E and eIF4G constitute the eIF4F complex that binds to the 5′ m7GTP-containing cap structure present on all cellular mRNAs. Initiation factor eIF4G also interacts with other factors, including eIF3 and eIF4B, to bring the mRNA to the ribosome for the initiation of translation (Clemens 2004). Overexpression of eIF4E is seen in a wide range of tumor types (Mamane and others 2004) and is associated with resistance to apoptosis (Ruggero and others 2004), whereas high-level expression of 4E-BP1 is proapoptotic (Li and others 2002). The latter protein is a key component in mediating the oncogenic effects of the Akt/Akt and extracellular-regulated kinase signaling pathways and integrates their function in tumors (She and others 2010).
We have shown previously that tumor necrosis factor (TNF) α–related apoptosis-inducing ligand (TRAIL) regulates the state of phosphorylation of 4E-BP1 and inhibits protein synthesis in malignant cell lines (Jeffrey and others 2006). TRAIL is a proapoptotic member of the TNF-α family that has preferential activity against malignant cells. The latter property gives this cytokine considerable clinical potential for selective anticancer therapy, although many tumors can become resistant to TRAIL (Johnstone and others 2008; Wang 2008). Death of TRAIL requires the binding of TRAIL to either of 2 receptors, TRAIL-R1 and TRAIL-R2 (MacFarlane 2003; Pennarun and others 2010; Dickens and others 2012). TRAIL–receptor interaction leads to formation of a death-inducing signaling complex (DISC) in a process that involves the recruitment of the adaptor protein Fas-associated death domain (FADD) and pro-caspase-8 (MacFarlane 2003; Dickens and others 2012). Within this complex, pro-caspase-8 then undergoes sequential proteolytic cleavages that result in its full activation (Chang and others 2003). The active caspase-8 dissociates from the DISC and initiates a cascade of activation of effector caspases that can in turn cleave a wide range of protein substrates, resulting in apoptosis. The activation of caspase-8 within the DISC is counteracted by the cellular FLICE-inhibitory protein (c-FLIP), which exists as at least 2 splice variants, namely, the short (c-FLIPS) and long (c-FLIPL) forms (Chang and others 2003; Kataoka 2005; Bagnoli and others 2010). In the case of c-FLIPL the protein can form a heterodimer with pro-caspase-8 and is itself partially processed. Both partially cleaved pro-caspase-8 and cleaved c-FLIPL are retained within the DISC and are not readily released into the cytosol (Chang and others 2003).
In this study we have focused on the effects of TRAIL on the mechanisms and pathways involved in the downregulation of translation that occurs prior to the onset of cell death in Jurkat T lymphoma cells. In other cell systems interferon-α (IFNα) has been shown to enhance the effects of TRAIL at the level of translational regulation (Jeffrey and others 2006). However, here we report a novel protective effect of IFNα on both the downregulation of translation and subsequent induction of apoptosis by TRAIL. We show that prior treatment of the cells with IFNα has a marked protective effect on protein synthesis, increasing by 10-fold the requirement for TRAIL for inhibition of translation and dephosphorylation of 4E-BP1. IFN treatment also blocks the dephosphorylation of Akt that normally occurs in response to TRAIL. However, inhibition of the IFN-regulatable PI3K pathway that lies upstream of Akt does not block the protective effect of IFN toward protein synthesis, suggesting an alternative mechanistic basis for modulation of the effects of TRAIL in Jurkat cells.
Materials and Methods
Materials
Materials for tissue culture were from Sigma-Aldrich, Inc. (Poole, United Kingdom) and Invitrogen Ltd. (Paisley, United Kingdom). Antibodies against the following proteins were from the sources indicated: total 4E-BP1, phosphorylated 4E-BP1 (Ser65, Thr37/46, and Thr70), and caspase-8 (Cell Signalling Technology, Hitchin, United Kingdom); FADD (BD Transduction Laboratories, Lexington, KY); TRAIL-R2 (Alexis Biochemicals, Exeter, United Kingdom); c-FLIP (Pharmingen, Oxford, United Kingdom and Merck Frosst Centre, Kirkland, Quebec, Canada); α-tubulin (Sigma-Aldrich, Inc.); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Advanced Immunochemical, Inc., Long Beach, CA). Hybond-P PVDF blotting membrane was from Sigma-Aldrich, Inc. TRAIL was from PeproTech EC Ltd. (London, United Kingdom) and IFNα2b (Intron A) from Schering-Plough Ltd. (Welwyn Garden City, United Kingdom). Biotinylated TRAIL was prepared and purified as previously described (Harper and MacFarlane 2008). The PI3K inhibitor LY294002 was from Calbiochem (Nottingham, United Kingdom) and all other chemicals were from Sigma-Aldrich, Inc.
Cell culture and cytokine treatments
Jurkat A3 human T-lymphoma cells were kindly provided by Professor S.J. Morley. The cells were grown at a density between 3×105 and 6×105 cells/mL. Where indicated the cells were preincubated without or with IFNα2b (500–1,000 U/mL) for 16–24 h. Unmodified or biotinylated TRAIL was then added for various times up to 4 h, at the concentrations stated in the figure legends. For the majority of the experiments recombinant TRAIL was added at concentrations within the range 0.5–50 ng/mL. When cells were treated with biotinylated TRAIL (for DISC analysis) a concentration of 500 ng/mL was used since this has been shown to be optimal for this modified form of the cytokine (Harper and MacFarlane 2008).
Determination of protein synthesis rates
Rates of overall protein synthesis in intact cells were measured by the incorporation of [35S]methionine (in the range of 1–10 μCi/mL) into trichloroacetic acid (TCA)–insoluble material during the last 60 min of the incubation. Radioactivity was determined as previously described (Jeffrey and others 2002). Measurements were performed with 3–6 replicates and rates of protein synthesis are expressed as counts per min incorporated per 105 cells. Independent assays were performed at least 3 times.
Caspase-8 assays
Cells were seeded at 4×105 cells/mL and incubated with or without IFNα and/or TRAIL as previously described. The cells were then lysed in caspase lysis buffer [10 mM HEPES (pH 7.3), 2 mM EDTA, 0.1% (v/v) NP-40, 5 mM dithiothreitol, 1 mM phenylmethanesulphonyl fluoride, 10 μg/mL pepstatin, 20 μg/mL leupeptin, and 10 μg/mL aprotinin] and incubated on ice for 10–15 min. Cytoplasmic extracts were made by centrifugation at 10,000 g for 10 min. Caspase-8 activity was determined in a Packard Fusion microplate reader, using the fluorogenic substrate acetyl-Ile-Glu-Thr-Asp-7-amino-4-methylcoumarin (Ac-IETD-AMC). Twenty microliters of cell extract were incubated with 200 μL of reaction buffer [100 mM HEPES (pH 7.3), 20% (v/v) glycerol, 0.5 mM EDTA, and 5 mM dithiothreitol] and 2 μL of substrate (5 mM). Reactions were incubated at 37°C for 1 h and the product was quantitated by fluorescence using an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Protein concentrations were determined and caspase activities were calculated as relative fluorescence units per μg protein.
Immunoblotting of cell extracts
Cells were harvested, washed in phosphate-buffered saline, and lysed as described previously (Jeffrey and others 2002). Protein concentrations were determined and samples containing equal amounts of protein were subjected to electrophoresis on sodium dodecyl sulfate (SDS) polyacrylamide gels. The proteins were transferred to PVDF membranes using a semi-dry blotting apparatus (Bio-Rad, Hemel Hempstead, United Kingdom). The blots were blocked, incubated with the appropriate primary antibodies, and developed using horseradish peroxidase-linked secondary antibodies. Enhanced chemiluminescence was performed using Lumiglo reagent (Cell Signalling Technology) according to the manufacturer's instructions. Levels of α-tubulin or GAPDH were determined as loading controls. All immunoblotting experiments were repeated at least two times and typical results are shown.
DISC isolation
Jurkat cells (6×107) were incubated with and without IFNα (1,000 U/mL) for 20 h and the volume of medium was subsequently reduced such that the cells were at a final concentration of 2×106/mL. Cells were exposed to 500 ng/mL of biotinylated TRAIL and immediately placed on ice for 1 h, followed by incubation at 37°C for 30 min. DISC complexes were then isolated by lysing the cells on ice for 30 min in DISC lysis buffer [30 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, and 1% (v/v) Triton X-100] in the presence of protease inhibitors (1 tablet of Complete Mini, EDTA-free protease inhibitor cocktail per 10 mL; Roche Diagnostics Ltd., Burgess Hill, United Kingdom). Lysates were then cleared by centrifuging at 15,000 g for 30 min and the biotinylated-TRAIL and its associated proteins were bound to streptavidin beads (Dynabeads M-280 Streptavidin; Invitrogen Dynal AS, Oslo, Norway) overnight. After washing in lysis buffer, the bound proteins were eluted with gel sample buffer and analyzed for DISC components by SDS gel electrophoresis and immunoblotting. The cell lysates from which the DISC complexes were isolated were analyzed in parallel for the same proteins.
Statistical analysis of data
The data for the effects of IFNα and TRAIL on inhibition of protein synthesis are representative of at least 3 independent experiments. Where indicated the results are the means±SEM of 3–6 determinations for each condition. Statistical significance of differences was determined by unpaired or paired t-tests, as appropriate, and P values are given in the legends to the figures.
Results
Effects of TRAIL and IFNα on regulation of protein synthesis in Jurkat cells
We have previously shown that, as part of the early cellular response to TRAIL in tumor cells, overall translation is strongly inhibited (Jeffrey and others 2006). In accordance with these earlier results, in Jurkat cells TRAIL inhibited the incorporation of [35S]methionine into total protein in a dose-dependent manner (Fig. 1A). In this cell type half-maximal inhibition occurred at a concentration of TRAIL between 1 and 5 ng/mL (Fig. 1B). The inhibition was not due to a loss of cell viability, which remained high for at least 4 h of TRAIL treatment (data not shown). Although IFNα alone exerted a small inhibitory effect on protein synthesis in these cells (probably due to induction of translational inhibitors such as the protein kinase PKR) (Garcia and others 2006) prior treatment of the cells with IFNα markedly impaired the effect of TRAIL. Thus, at concentrations of TRAIL of 5 ng/mL and above the rate of overall protein synthesis was consistently higher in cells pretreated with IFNα than in cells not exposed to this agent (statistically significant, P<0.01) (Fig. 1A). Moreover, in the presence of IFNα at least a 10-fold higher concentration of TRAIL (50 ng/mL) was required to inhibit protein synthesis by 50% (Fig. 1B). Similar inhibitory effects of IFNα were observed when the effects of TRAIL on apoptosis were examined. There was a decrease in cells with a sub-G1 DNA content from 11.8% without IFNα to 8.1% with IFNα, following treatment with 0.5 ng/mL of TRAIL. In cells treated with 5 ng/mL of TRAIL there was a decrease from 25.6% without IFNα to 13.5% with IFNα (Supplementary Fig. S1A; Supplementary Data are available online at
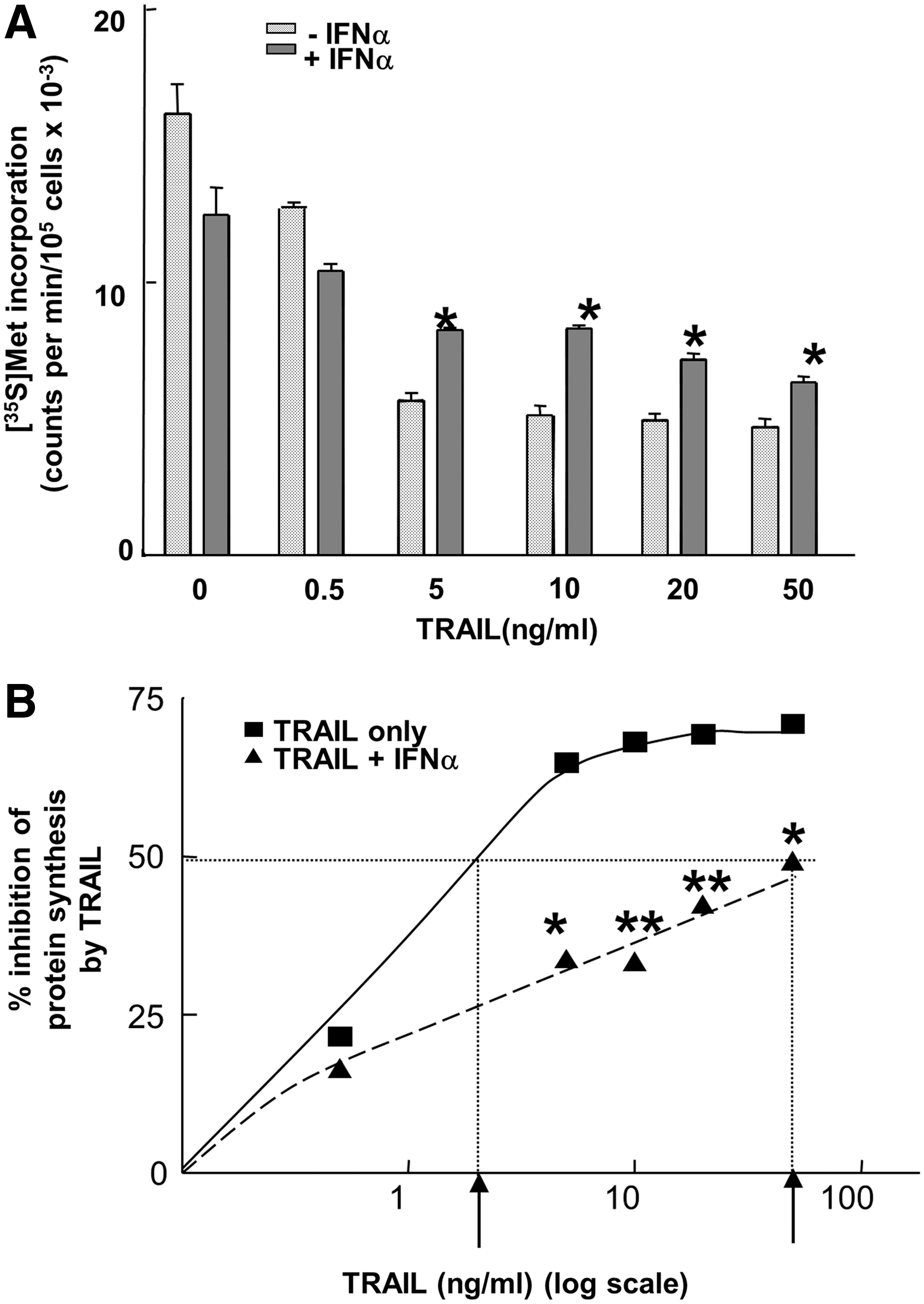
Effect of interferon-α (IFNα) on the sensitivity of protein synthesis to inhibition by tumor necrosis factor α–related apoptosis-inducing ligand (TRAIL) in Jurkat cells. Jurkat cells were seeded at 4×105 cells/mL and incubated in the absence or presence of human IFNα (1,000 U/mL) for 24 h. The cells were then treated with TRAIL at the concentrations indicated for 4 h.
The inhibition of protein synthesis by TRAIL was associated with marked dephosphorylation of the eIF4E binding protein 4E-BP1, as shown in Figure 2. A shift from the most highly phosphorylated (γ) to the least phosphorylated (α) form of the protein was seen with increasing concentrations of TRAIL. Moreover, immunoblotting with antibodies to specific phosphorylation sites on 4E-BP1 revealed major decreases in the levels of phosphorylation at residues Thr37/46, Ser65, and Thr70. The most noticeable decrease involved Ser65, with exposure of the cells to as little as 0.5 ng/mL of TRAIL resulting in marked loss of phosphorylation at this site. When the cells were pretreated with IFNα, TRAIL at 0.5 ng/mL had no effect on the phosphorylation of Ser65 and a 10-fold higher concentration was needed to produce significant dephosphorylation. These results are concordant with the protective action of IFNα toward the effects of TRAIL on protein synthesis in Jurkat cells.

Effects of IFNα and TRAIL on the phosphorylation state of 4E-BP1. Jurkat cells were incubated in the absence or presence of IFNα (1,000 U/mL) for 16 h. The cells were then treated with TRAIL at the concentrations indicated for 4 h. Extracts were prepared and analyzed by gel electrophoresis and immunoblotting using antibodies against total 4E-BP1 (top panel) and phosphospecific antibodies against Thr37/46, Ser65, and Thr70 of 4E-BP1 (as indicated). The extracts were also immunoblotted for α-tubulin as a loading control (bottom panel). The differentially phosphorylated α, β, and γ forms of 4E-BP1 are indicated in the top panel.
It has previously been demonstrated that IFN treatment stimulates the phosphorylation of 4E-BP1 by a mechanism that depends on the activity of the PI3K-Akt-mTOR axis (Lekmine and others 2003). We therefore investigated the role of PI3K in regulating protein synthesis in Jurkat cells by blocking the activity of the enzyme with the PI3K inhibitor LY294002. Exposure of Jurkat cells to this compound alone at a concentration of 10 μM inhibited protein synthesis by 48% (statistically significant, P=0.0001) (Fig. 3A). Moreover, LY294002 enhanced the inhibitory effect of a low concentration of TRAIL (0.5 ng/mL) from 15.3% to 26.6% inhibition (on top of the effect of LY alone) (statistically significant, P<0.05; Fig. 3B). However, the more dramatic inhibition of protein synthesis brought about by exposure to a 10-fold higher concentration of TRAIL was not enhanced by LY294002 (Fig. 3B).
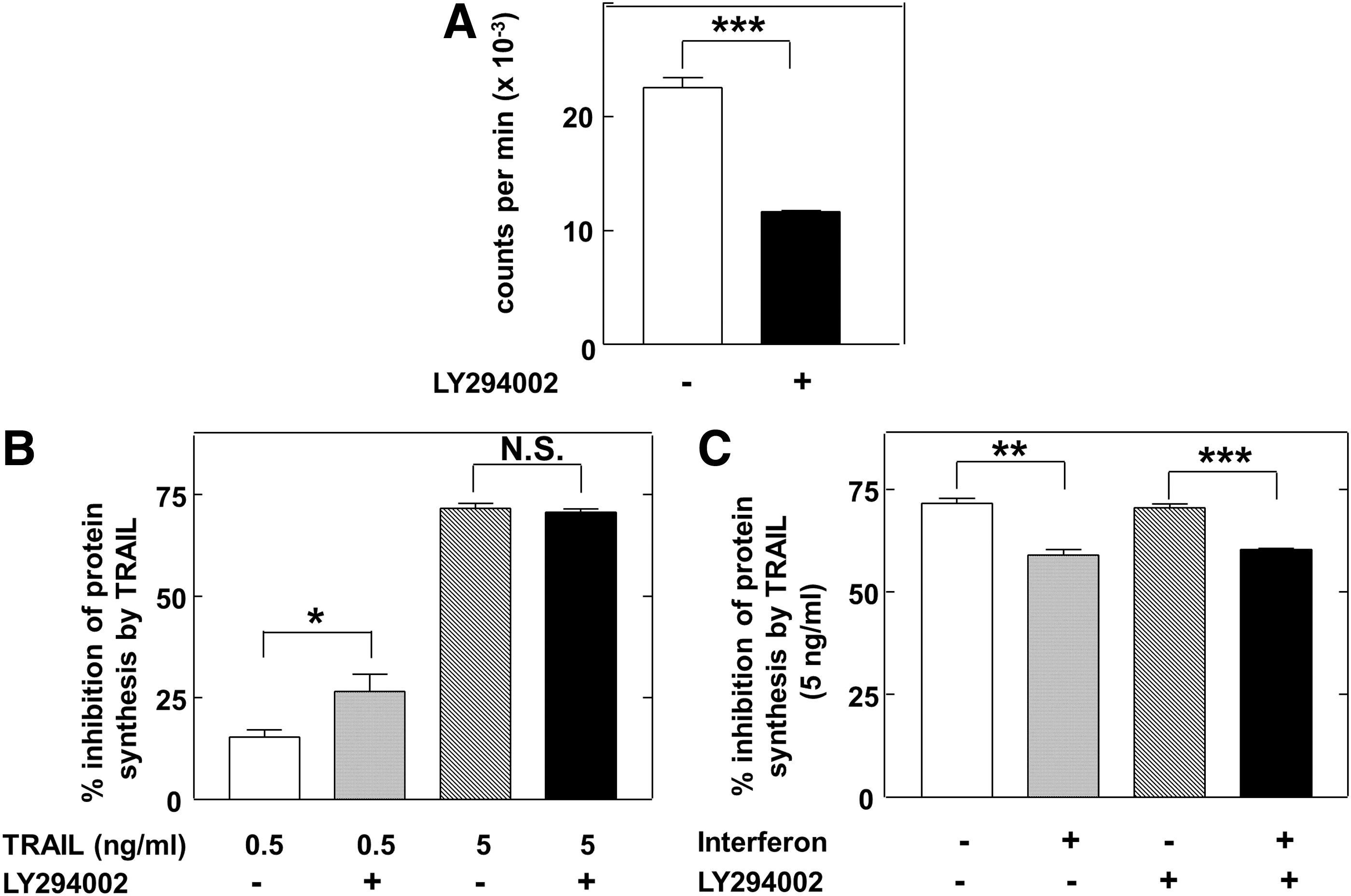
Effects of LY294002 on protein synthesis and its regulation by TRAIL and IFNα.
Since IFNα has been shown to activate PI3K (Kaur and others 2005) we also examined the effect of blocking PI3K activity on the ability of IFN to impair the inhibition of protein synthesis by TRAIL. Figure 3C shows that LY294002 did not prevent this effect of IFN since there were similar, statistically significant, effects of the latter both in the absence and presence of the inhibitor. This suggests that activation of PI3K is not involved in the mechanism by which IFN protects Jurkat cells from the effects of TRAIL. Nevertheless, IFN was able to block the TRAIL-induced dephosphorylation of the Akt at position Ser473 (Fig. 4A), perhaps indicating that there are additional, PI3K-independent, pathway(s) regulating the phosphorylation of Akt in Jurkat cells. Consistent with this, LY294002 alone did not cause major dephosphorylation of Akt at Ser473, although it enhanced the effect of TRAIL (Fig. 4B). Such additional pathways may constitute the targets for TRAIL and IFN action.
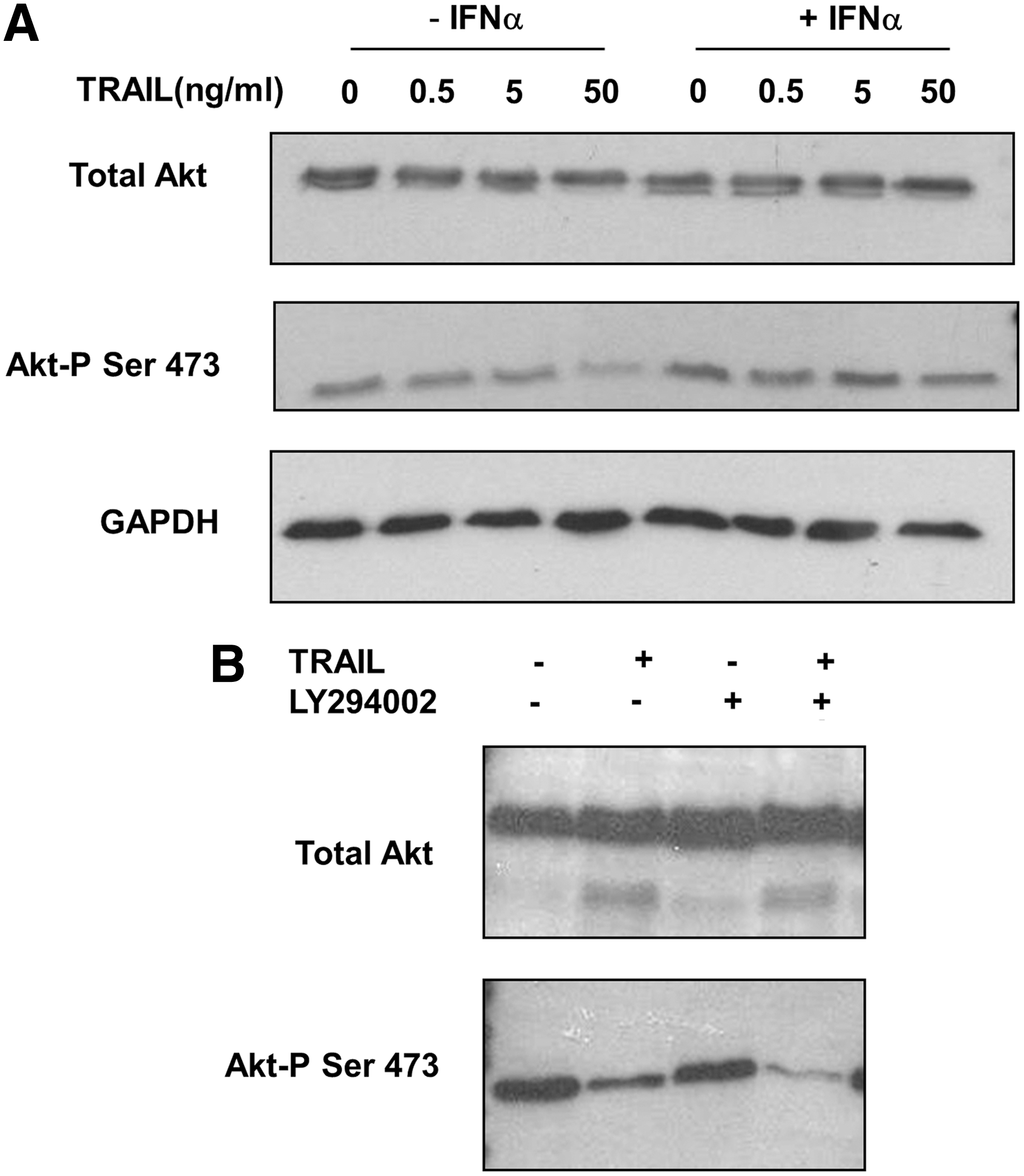
Effects of IFNα, TRAIL, and LY294002 on the phosphorylation of protein kinase B (Akt).
We have analyzed some of the initial events that instigate TRAIL-induced apoptosis to determine whether they are also impaired by prior exposure of Jurkat cells to IFNα. IFNα had no significant effect on the levels of FADD or pro-caspase-8, which are key constituents of the TRAIL DISC (Fig. 5A). IFNα also did not affect the expression of the TRAIL receptor R2, as measured by flow cytometry (which also showed that very little TRAIL-R1 is detectable in Jurkat cells—data not shown). Further, treatment of Jurkat cells with IFNα did not significantly alter the levels of either the long or short forms of the potential caspase-8 antagonist c-FLIP (Fig. 5A). To determine whether apical events at the level of the TRAIL DISC itself were affected by IFN treatment, we isolated this complex after treating cells for 30 min with biotinylated TRAIL and subjected the extracts to affinity chromatography on streptavidin-linked magnetic beads. Previous studies have shown that in this system TRAIL DISC formation is rapid, reaching a peak in Jurkat cells at 30 min (Harper and MacFarlane 2008). Figure 5B shows that IFNα had no effect on the TRAIL-mediated recruitment of the receptor TRAIL-R2, indicating that similar levels of the TRAIL DISC can be isolated from cells pretreated or not with IFNα. IFN did not affect the level of FADD associated with the DISC. However, there was a slight increase in unprocessed full-length pro-caspase-8 within the TRAIL DISC in IFNα-treated cells (Fig. 5B). Consistent with this, the processing of caspase-8 to generate the catalytically active p18 subunit of the enzyme was partially impaired by IFN treatment of Jurkat cells (Fig. 5C) (this subunit is rapidly released from the DISC and cannot easily be observed in the complex.). These findings suggest that TRAIL-dependent formation of the DISC is not compromised by IFN treatment but that the subsequent activation and processing of caspase-8 is partially inhibited. The conclusion that full TRAIL-induced activation of caspase-8 is compromised in IFN-treated Jurkat cells is supported by the observation that, although total cytoplasmic levels of the caspase-8 substrate c-FLIPL were unchanged, there was less cleavage of this protein to its p43 form within the DISC (Fig. 5B, bottom panel) and reduced generation of the p43 form in the total cytoplasmic fraction (Fig. 5D) in cells pretreated with IFNα. The changes in levels of caspase-8 p18, caspase-8 p41/43, and c-FLIP p43 were consistent between multiple experiments. The impairment of caspase-8 activation was confirmed by assays of the activity of this enzyme using a fluorometric substrate, Ac-IETD-AMC (Fig. 5E). Although caspase-8 activity was inhibited in IFN-treated cells there were no changes in the levels of the potential caspase-inhibitory proteins cIAP1, cIAP2, or XIAP in response to either IFNα or TRAIL treatment (Supplementary Fig. S2). These data suggest that the attenuation by IFNα of the TRAIL-mediated inhibition of overall protein synthesis and subsequent apoptosis in Jurkat cells is associated with downregulation of caspase-8 activation within the TRAIL DISC rather than inhibition of cytoplasmic caspase-8 activity by inhibitor of apoptosis proteins (IAPs).
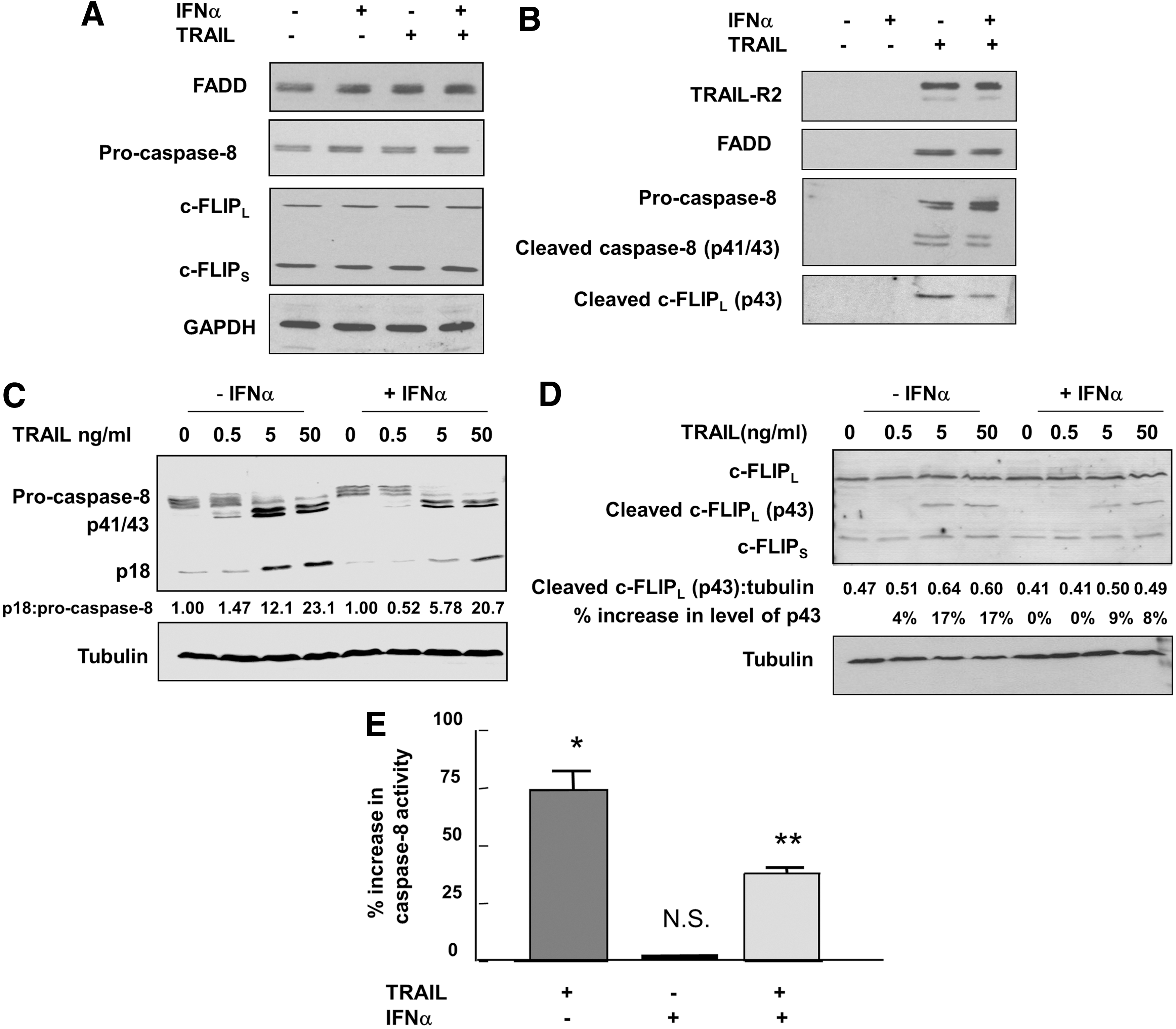
Effects of IFNα and TRAIL on key components of the TRAIL death-inducing signaling complex (DISC) and activation of caspase-8 in Jurkat cells. Cells were incubated in the absence or presence of human IFNα2b (1,000 U/mL) for 20 h and then treated with biotinylated TRAIL (500 ng/mL) for 30 min.
Discussion
Exposure of cells to proapoptotic stimuli results in early inhibition of protein synthesis (Tee and Proud 2000; Jeffrey and others 2002; Constantinou and Clemens 2005). Here we have shown that such inhibition, and the extensive dephosphorylation of 4E-BP1 that accompanies it in response to TRAIL, can be inhibited by pretreatment of Jurkat cells with IFNα. In some tumor cell types IFN treatment leads to stronger proapoptotic effects of TRAIL (Chawla-Sarkar and others 2002; Clark and others 2010) and previously we reported a synergistic inhibition of the rate of protein synthesis by IFNα and TRAIL in MCF-7 breast cancer and Hela cells (Jeffrey and others 2006). However, in other cell types, particularly those of lymphoid or myeloid origin, IFN treatment protects against apoptosis (Chaouchi and others 1994; Jewell and others 1994; Liu and others 1999) and renders the cells less sensitive to TRAIL [reviewed in Clemens (2003)].
PI3K activity provides antiapoptotic signals in tumor cells, largely as a result of the activation of Akt (Downward 2004). PI3K and Akt are constitutively active in Jurkat cells, due to a mutation of the negative regulatory phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene (Shan and others 2000), and most likely contribute to the activation of protein synthesis since Akt is well established as an activator of mTOR (Mamane and others 2006). We have shown that the PI3K inhibitor LY294002 strongly inhibits protein synthesis and also enhances the effect of a suboptimal concentration of TRAIL (Fig. 3A, B). This is consistent with the reported ability of Akt to counteract the effects of TRAIL in prostate cancer cells (Nesterov and others 2001). The IFNs have been shown to activate PI3K, Akt, and mTOR signaling in other systems (Kaur and others 2005, 2012) but it is unlikely that this effect contributes to the desensitization of Jurkat cells to TRAIL because of the constitutive activation of PI3K. Consistent with this, IFNα did not enhance the phosphorylation of 4E-BP1 in Jurkat cells (Fig. 2), contrary to its effects in U-266 or KT1 cells (Lekmine and others 2003; Kaur and others 2007). Moreover, IFNα was still able to exert a protective effect against the downregulation of protein synthesis by TRAIL in the presence of LY294002 (Fig. 3B). IFN did prevent the TRAIL-induced dephosphorylation of Akt at Ser473 (Fig. 4A) but this apparent anomaly can be explained by the existence of alternative pathways for the phosphorylation and activation of Akt (Mahajan and Mahajan 2012), a conclusion that is supported by the insensitivity of the phosphorylation of both Akt (Fig. 4B) and 4E-BP1 (data not shown) to LY294002 in Jurkat cells. Such alternative pathways may be the targets for the opposing effects of TRAIL and IFNα.
In cell types that are sensitized to TRAIL by prior IFN treatment, the level of expression of pro-caspase-8 has been shown to be upregulated (Kim and others 2002; Fulda and Debatin 2006; Liedtke and others 2006). In contrast, there is little, if any, change in pro-caspase-8 expression in IFN-treated Jurkat cells (Fig. 5A, C). Further, although enhancement of c-FLIP levels by IFNγ has previously been reported (Grassi and others 2004), neither c-FLIPL nor c-FLIPS is significantly upregulated by IFNα in Jurkat cells (Fig. 5A, D). The same is true of the caspase inhibitors cIAP1, cIAP2, and XIAP. The antagonistic effect of IFNα toward the actions of TRAIL in Jurkat cells therefore cannot be explained by any overall increase in c-FLIP or IAP expression. Although the level of total c-FLIPL is not affected there is reduced cleavage of this protein within the TRAIL DISC (Fig. 5B). This is most likely a reflection of the impaired activity of caspase-8, which is responsible for c-FLIPL cleavage within the DISC (Kataoka 2005; Dickens and others 2012), in cells that have been pretreated with IFNα. Other aspects of TRAIL-mediated DISC formation, such as recruitment of the TRAIL receptor TRAIL-R2 or the adaptor protein FADD, were unaffected by IFN treatment (Fig. 5B).
Several reports have shown that IFNs can protect lymphoid cells from apoptosis (Clemens 2003) but the molecular mechanisms responsible for such effects are not well established. Activation of PI3K and its downstream target mTOR has been implicated in the type-I IFN-induced survival of primary B cells (Ruuth and others 2001) and may counteract the effects of TRAIL by inhibiting the expression of proapoptotic and tumor-suppressive proteins (Kaur and others 2007). However the constitutive activation of the PI3K/Akt/mTOR pathway in Jurkat cells leaves relatively little scope for a further IFN-induced increase in this signal transduction mechanism. An alternative mechanism for the protective effect of IFNα may involve the activation of the NFκB pathway (Yang and others 2005), which can induce the expression of antiapoptotic proteins such as Mcl-1 (Jourdan and others 2000) and cause TRAIL resistance (Henson and others 2003). IFNα also induces the survival factor G1P3 (ISG 6–16), which antagonizes TRAIL effects in myeloma cells (Cheriyath and others 2007), but it is not known whether Mcl-1 or G1P3 has any direct effect on translation.
We propose that IFNα modulates the control of protein synthesis by impairing the process of caspase-8 activation by TRAIL. The mechanism of this effect could involve induction of novel components of the DISC that regulate caspase-8. For example, it has been shown that the heat shock protein 90 (HSP90) can recruit c-FLIPS to the TRAIL DISC and that this can contribute to resistance to TRAIL (Panner and others 2007). The expression of HSP90 is enhanced by IFNα in some systems (Caraglia and others 1999), but examination of this possible mechanism in Jurkat cells requires further study. Another possible route by which IFN treatment could protect cells against TRAIL-mediated inhibition of protein synthesis and induction of apoptosis may involve the stimulation of autophagy. Several recent reports have shown that IFNs can induce autophagy and that this counteracts apoptosis in tumor cells (Ambjorn and others 2013; Schmeisser and others 2013; Zhu and others 2013). Further, the formation of the TRAIL DISC is enhanced when autophagy is suppressed (He and others 2012). It will be of interest in future work to determine whether the protection of Jurkat cells by IFNα against the various effects of TRAIL is mediated by enhanced autophagy.
A prosurvival effect of IFNα toward cells of lymphoid or myeloid lineages may be physiologically important in allowing the immune system to elicit adaptive defense responses to viral and other infections. For the overall survival of the organism this may be more critical than the antigrowth or antitumor effects of the IFNs that are often observed in non-lymphoid cell types. Nevertheless, tumors and tumor cell lines of lymphoid origin can undergo apoptosis in response to IFN (Huang and others 2007), and in some cases this may be TRAIL mediated (Oshima and others 2001). Indeed, IFNα has been shown to induce TRAIL in lymphoid cells (Toomey and others 2001; Ghosh and others 2003; Yanase and others 2005). However, our results suggest that, whereas the combination of TRAIL and IFNα may have significant potential for the treatment of many malignancies, such a strategy might be inappropriate (or even counter-productive) for certain types of lymphoid tumors.
Footnotes
Acknowledgments
The authors thank Professor S.J. Morley for the Jurkat cells. This work was supported by grants from the Wellcome Trust, the Ralph Bates Pancreatic Cancer Research Fund, and the Medical Research Council.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.