
Abstract
Endothelial progenitor cells (EPCs) may contribute to vascular repair and angiogenesis. Chemokine (C-X-C motif) ligand 12 (CXCL12/SDF-1) is known to play an important role in the mobilization and recruitment of progenitor cells. Therefore, we assessed the function of CXCL12 as a stimulating molecule of angiogenesis in EPCs and the underlying mechanism after intracerebral hemorrhage (ICH). Isolated EPCs from male Sprague-Dawley rats, stimulate with various doses of CXCL12. Then, 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was used to assess the proliferation of EPCs, and cell migration and adhesion were analyzed by transwell chamber assay. Furthermore, mRNA levels of endothelial markers von Willebrand Factor (vWF), Tie-2, and vascular endothelial cadherin (VE-cadherin) were explored by real-time polymerase chain reaction. Capillary tube and vessel formation in vitro and in vivo were detected after pretreatment with the C-X-C chemokine receptor type 4 (CXCR4) inhibitor AMD3100. Following stimulation with various doses of CXCL12, an obvious dose-dependent increase in the proliferation, migration, and adhesion of EPCs was confirmed. Furthermore, the mRNA levels of endothelial markers vWF, Tie-2, and VE-cadherin were also demonstrated in CXCL12-treated EPCs, indicating that CXCL12 could regulate EPC differentiation to endothelial cells. Importantly, these increases depended on the activation of CXCR4 signaling, as pretreatment with CXCR4 inhibitor AMD3100 dramatically dampened the CXCL12-induced effects. Additionally, blocking CXCR4 signaling dampened CXCL12-induced angiogenic activity both in vitro and in vivo. Following construction of a rodent ICH model, scaffolds delivering CXCL12 together with EPCs resulted in an evident increase in blood vessel formation; however, this increase in blood vessels was attenuated with delivery of AMD3100. CXCL12 stimulates EPCs to induce angiogenesis though the CXCR4 pathway after ICH. Consequently, our findings provide a potential target for angiogenesis in ICH.
Introduction
H
The endothelium is an essential component of the cardiovascular system, playing a vital role in blood vessel formation, vascular homeostasis, permeability, and the regulation of inflammation (Kirton and Xu 2010). Endothelial progenitor cells (EPCs) are a group of pluripotent cells and are mainly derived from embryonic mesoderm and postnatal bone marrow, primarily in the umbilical cord blood and peripheral blood circulation (Pearson 2010). EPCs are known to have the potential to differentiate into endothelial cells (ECs). EPCs are involved in normal, pathological, and pathophysiological neovascularization, such as embryonic development, tumor growth, and wound healing (Park and others 2011). Furthermore, several reports have demonstrated that EPCs can not only participate in fetal blood vessel formation, but can also be mobilized to the site of injury after birth to trigger vascular repair and regeneration (Ebrahimi and others 2012; Kim and others 2012). However, the effect of EPCs on vascular repair is limited by the number of EPCs after birth. A report shows that pretreatment with estrogen can enhance the proliferation and differentiation of EPCs (Matsubara and Matsubara 2012). It is suggested that searching for new molecules that can regulate EPC proliferation and differentiation, and promote angiogenesis will be an important target in the therapy of ICH.
Chemokine (C-X-C motif) ligand 12 (CXCL12) is a small pro-inflammatory chemoattractant cytokine that was first cloned by Tashiro and others as a protein captured by its specific amino-terminal signal sequences (Tseng and others 2011). It was later identified functionally as a homeostatic chemokine whose major function is to regulate hematopoietic cell trafficking (Teicher and Fricker 2010). Nowadays, CXCL12 is considered to play an important role in the mobilization and recruitment of progenitor cells to the neoangiogenic niches, supporting revascularization of ischemic tissue and tumor growth (Kioi and others 2010). Proliferation of ECs is the latent condition of angiogenesis, and researches in EPCs or stem cell transplantation of ischemic disease have shown that high expression of CXCL12 in ischemic tissue is a significant factor involved in stem/progenitor cell homing (Vagima and others 2011). It is suggested that CXCL12 may play an important role in the recruitment of EPCs. Therefore, investigation of the effect of CXCL12 on EPCs may provide a potential target for angiogenesis.
In this study, we aimed to assess the effect of CXCL12 on angiogenesis based on EPCs. After stimulation with CXCL12, the proliferation, migration, adhesion, differentiation into ECs, and the formation of microtubules were analyzed. Furthermore, the pro-angiogenic activity of CXCL12 was explored in an animal model of ICH, and the underlying mechanism was also analyzed.
Materials and Methods
Antibodies and reagents
If not otherwise mentioned, all substances were purchased from Gibco (Grand Island, NY). Lymphocyte separation medium was bought from Abcam (Cambridge, MA). AMD3100 (C-X-C chemokine receptor type 4, CXCR4 inhibitor) was bought from Calbiochem (San Diego, CA). Rabbit anti-CXCR4 polyclonal antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). β-Actin antibodies were obtained from Upstate Biotechnology, Inc. (Lake Placid, NY). Rabbit anti von Willebrand Factor (vWF) antibodies were purchased from Abcam (Cambridge, MA). Anti-Tie-2 antibodies were bought from Sigma (St. Louis, MO). The monoclonal antibody against vascular endothelial cadherin (VE-cadherin) was obtained from Cell Signaling Technology (Beverly, MA). HRP-conjugated goat anti-mouse and anti-rabbit antibodies were from Dako Cytomation (Glostrup, Denmark). Collagenase VII-S was bought from Sigma.
Isolation of EPCs
EPCs were obtained from male Sprague-Dawley (SD) rat (14–15 weeks old; Charles River Laboratories, Wilmington, MA) bone marrow in the following manner. SD rats were sacrificed by an overdose of anesthetic, then the tibia and femur were removed, and gently rinsed with 0.01 M phosphate-buffered saline (PBS) until the bone marrow scattered into a single-cell suspension. The suspension was carefully added to an equal volume of lymphocyte separation medium. After 2,000 rpm centrifugation for 20 min, the cells were washed with PBS 3 times, followed by collection of the cells.
EPCs culture and characterization
EPCs from SD rats were counted and diluted to 4×106 cells/mL, maintained in M199 medium supplemented with 20% FBS, 50 ng/mL VEGF, 1 ng/mL bFGF, and 2 ng/mL IGF-1, and then added to human fibronectin (1 ng/μL, 800 μL)-coated 6-well plates, 2 mL in each well. All cells were cultured at 37°C in a 5% CO2 incubator (Life Technologies, Baltimore, MD). After 7 days, Percent marker CD34 and CD133 positivity were determined using FACS analysis, 5,000 cells of each burette was quantitatived. Then, the cells were labeled with 1,1-dioctadecyl-3,3,3,3-tetramethylindocarbocyanine-labeled acetylated low-density lipoprotein (Dil-acLDL; Sigma) and FITC-labeled Ulex lectin I (FITC-UEA-I; Sigma), and they were examined with a laser scanning confocal microscope. Only double-positive cells were identified as EPCs.
MTT assay
EPCs were seeded into 96-well culture plates. After 7 days, the cells were treated with CXCR4 inhibitor AMD3100, or not, prior to exposure to doses of CXCL12 (0, 20, 40, 80, and 160 ng/mL). The cells were incubated for 24 h at 37°C and 5% CO2, then, the culture medium was replaced with fresh medium containing 5 ng/mL 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and the cells were incubated for an additional 4 h. Finally, formazan crystals were dissolved with a SDS-HCl solution and optical density was determined with a spectrophotometer at λ 490 nm.
Transwell migration assay
Transwell assays were performed with 8-μm porous chambers coated with 10 μL/mL fibronectin on the top and bottom according to the manufacturer's recommendations. About 25 μL of M199 medium consisting of control or test compounds was placed in the bottom well of the chamber. Cells were allowed to migrate through the membrane for 24 h. The cells that migrated to the lower surface of the membrane were then fixed and stained using a Giemsa kit, then counted under 5 fields (×200).
Adhesion assay
Human fibronectin (1 ng/μL) was used to coat 12-well culture dishes, after which the 7 days EPCs were incubated at a density of 1×105 cells/mL in their corresponding medium. After 30 min at 37°C and 5% CO2, the plate was washed twice with PBS, and the number of the adherent cells was counted. For cell counting, 0.05% trypsin/EDTA was added for 5 min at 37°C, and dissociation into a single-cell suspension was achieved by pipetting. The cells were subsequently resuspended in the basal medium. The number of cells was then determined under 5 fields (×200).
Real-time polymerase chain reaction
After EPCs were exposed to CXCL12/AMD3100 for 24 h, total cellular RNA was extracted using Unizol Reagent according to the manufacturer's instructions (Biostar, Shanghai, China). Two micrograms of RNA from each sample was used as a template for cDNA synthesis with a reverse transcription kit (Fermentas, St. Leon-Rot, Germany). An equal volume of cRNA product was used in the polymerase chain reaction (PCR). The primers used were as follows: vWF, 5′-GCTTGAACTGTTTGACGGAGAGG-3′ (forward primer) and 5′-TGACCCAGCAGCAGGATGAC-3′ (reverse primer); Tie-2, 5′-TCTGTGCTGTTCCTTCTTGC-3′ (forward primer) and 5′-CTTAGGTAACTTCCAGCGGA-3′ (reverse primer); VE-cadherin, 5′-TCCTCTGCATCCTCACTATCACA-3′ (forward primer) and 5′-GTAAGTGACCAACTGCTCGTGAAT-3′ (reverse primer); β-actin, 5′-GCACCACACCTTCTACAATG-3′ (forward primer) and 5′-TGCTTGCTGATCCACATCTG-3′ (reverse primer). The steps of quantificational real-time polymerase chain reaction (qRT-PCR) were performed as follows: 94°C for 2 min for initial denaturation; 94°C for 20 s, 58°C for 15 s, and 72°C for 15 s; 2 s for plate reading for 40 cycles; and melt curve from 65 to 95°C. β-Actin was used as a control to normalize the gene expression. PCR products were resolved on 2% agarose gels with 0.5 μg/mL ethidium bromide, visualized, and photographed.
Western blotting analysis
Cells were homogenized and lysed with RIPA lysis buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% Triton X-100, 1 mM EDTA, 10 mM beta-glycerophosphate, 2 mM sodium vanadate, and protease inhibitor). Protein concentration was assayed using the micro-BCA protein assay (Pierce, Rockford, IL). 40 μg of protein per lane was separated by 12% SDS-PAGE and electroblotted onto a polyvinylidene difluoride (PVDF) membrane in a semi-dry trans-blot apparatus. Then, the PVDF membrane was blocked by incubating with 5% nonfat milk in Tris Buffered Saline with Tween 20 (TBST) buffer at room temperature for 1 h. The PVDF membranes were respectively incubated with anti-CXCR4, anti-vWF, anti-Tie-2, anti-VE-cadherin, or anti-β-actin antibodies for 1 h at room temperature. Following 3 washes with TBST buffer, HRP-conjugated secondary antibodies were introduced, and enhanced chemiluminescence (ECL, Amersham Pharmacia, NJ) was used for detection.
Flow cytometry analysis of surface CXCR4 expression
Surface CXCR4 expression was analyzed by Flow cytometry. Cells were stained with rabbit anti-CXCR4 antibody followed by Alexa Fluor 594 goat anti-rabbit IgG (Molecular Probes, Eugene, OR). Surface CXCR4 expression was determined by an Epics Altra flow cytometer (Beckman Coulter, Fullerton, CA).
Tube formation assay
A Matrigel tube formation assay was performed to assess the ability of EPCs to integrate into a vascular structure as described (Arnaoutova and Kleinman 2010), A 48-well culture plate was coated with Matrigel plus 100 ng/mL VEGF. EPCs cultured with test compounds were seeded in the Matrigel-coated plate (5×104 cells/well). After 20 h, the vascular structural tube formed by EPCs was fixed and stained with the Diff-Quik® staining set (DADE BEHRING, Inc., Newark, DE), according to the manufacturer's instructions. After washing thrice with PBS, 4 areas of each sample were photographed under an inverted light microscope, and the number of tube branches was counted in triplicate.
Matrigel plug assay and immunostaining
The Matrigel plug assay was adapted from methods in the literature (Sahoo and others 2011). Nine C57BL/6 mice (8 weeks old) were subcutaneously injected in the abdomen with Matrigel plugs containing the test compounds. After implantation for 7 days, the mice were anesthetized, and the Matrigel plug was harvested along with the overlying skin and peritoneal membrane. Then, the harvested material was fixed in 4% paraformaldehyde and embedded in paraffin, and sections were stained with immunofluorescent anti-CD31 antibody (1:1,000) to observe vascularization. The microvessels were visualized under an FV1000 Olympus confocal microscope and counted in 9 random areas independent by 3 individuals. This protocol conformed to the guidelines of the Institutional Animal Care and Use Committee.
Animal model of ICH
All experiments were performed following an institutionally approved protocol in accordance with National Institutes of Health guidelines. ICH was induced by collagenase type VII injections. Male SD rats (Charles River Laboratories, Wilmington, MA) were anesthetized with isoflurane (1% to 1.2%) in 30%/70% oxygen/nitrous oxide. A catheter in the right femoral artery was used to measure blood pressure, pH, PaO2, and PaCO2. Using a stereotactic frame, 1 μL of saline containing 0.5 U collagenase VII-S was injected through a 22-gauge needle over 5 min into the right caudate nucleus. Ten rats per group were used.
EPCs were suspended in PBS at 3×106/mL. Thirty-six ICH rats were randomly divided into 4 experimental groups; each group consisted of 9 rats. Group 1 (G1) received only delivery of 1 mL PBS into the tail vein. Group 2 (G2) received delivery of 3×106 EPCs into the tail vein. Groups 3 (G3) and 4 (G4) received delivery of CXCL12 (10 μL, every other day) and 3×106 EPCs into the tail vein. From the second day, Group 4 animals received twice-daily subcutaneous injections of 120 μg/kg AMD3100. All treatments were started at 24 h after ICH and continued for 14 consecutive days.
Analysis of blood vessel ingrowth
The rats' brains were perfused with PBS, and then with 4% paraformaldehyde for 30 min under physiological pressure. The brain region of hemorrhage was isolated and fixed with 4% paraformaldehyde for 24 h, embedded in paraffin, and cut into 5-μm serial sections. The rat brain slices were incubated with anti-vWF primary antibodies (1:200), and then HRP-conjugated secondary antibodies were added. The blood vessels that were immunostained with vWF were determined manually at ×100 magnification in the total implant area and normalized to the implant area. All images were visualized using a Nikon Eclipse E800 light microscope and a Spot RT digital camera (Sterling Heights, MI).
Data analysis
Each experiment was repeated at least 3 times. The results are presented as mean±standard error of the mean. Statistical analyses were performed with a paired or unpaired Student t-test for direct 2-group comparisons and the Tukey–Kramer test after a significant 1-way analysis of variance F test for multiple-group comparisons. A difference was considered significant with P≤0.05.
Result
Identification of EPCs
The purity of the isolated EPCs were characterized by flow cytometry analysis (Fig. 1A). To indentify EPCs, FITC-UEA-I, and Dil-acLDL were used to label the cells. Laser scanning confocal microscopy analysis showed large numbers of double-positive cells, indicating that EPCs had been successfully isolated (Fig. 1B).
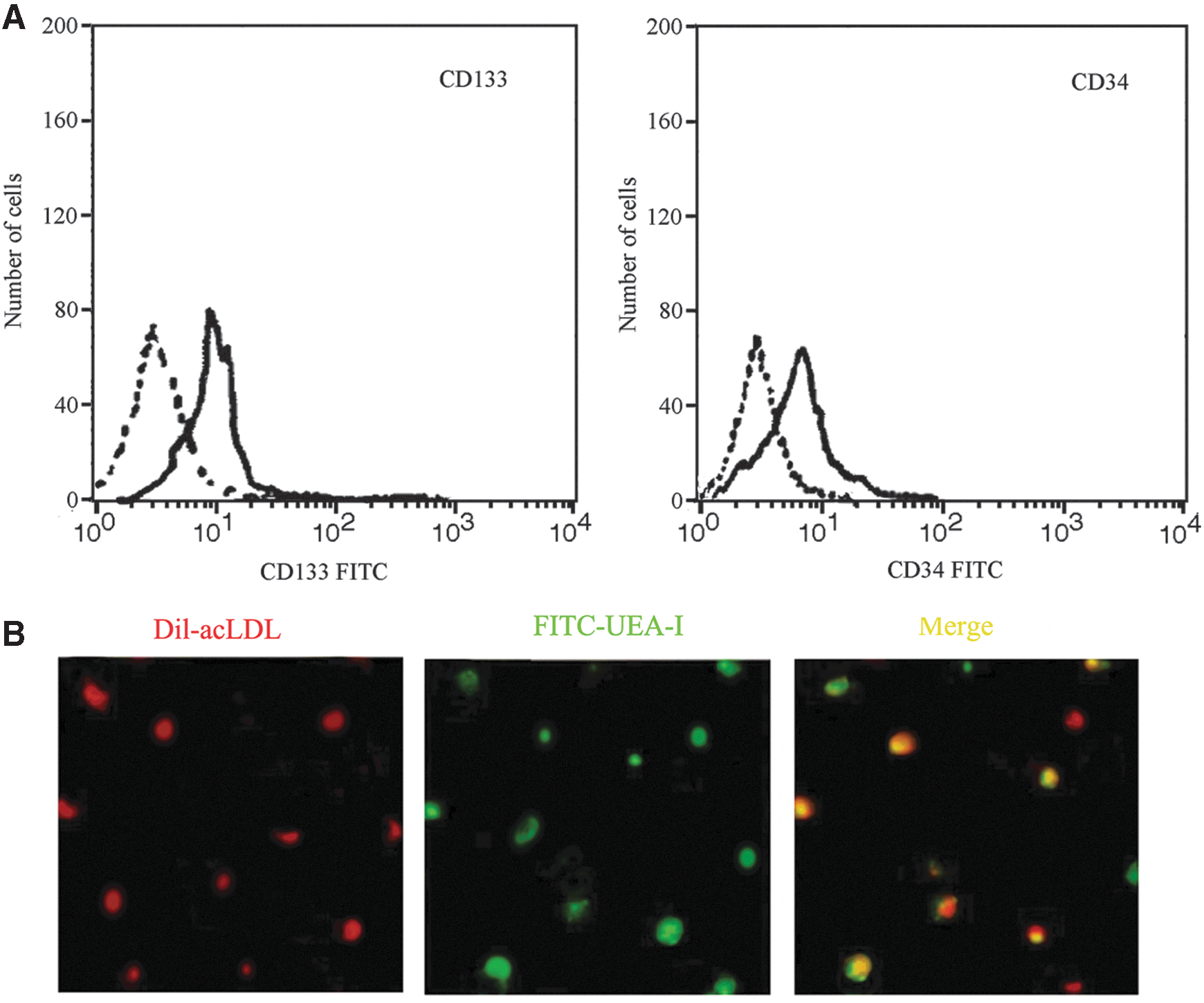
Characterization of EPCs by flow cytometry and immunofluorescence. In flow cytometry analysis, the purity of the isolated EPCs was delimited by CD34 and CD133
CXCL12 induced EPC proliferation, migration, and adhesion in a dose-dependent manner
To explore the effect of CXCL12 on EPC proliferation, the MTT assay was used. After stimulated with different concentrations of CXCL12 for 24 h, an obvious increase in cell proliferation rate was observed compared with control groups, and reached a peak when pretreated with 80 ng/mL CXCL12 (Fig. 2A). These results indicated that CXCL12 dose-dependently enhanced EPC proliferation.

CXCL12 induced proliferation, migration, and adhesion of EPCs. EPCs were treated with various doses of CXCL12, then cell proliferation
We further analyzed whether CXCL12 pretreatment could influence the chemotaxis of EPCs. The dicated dose of CXCL12 was introduced in a transwell migration assay. As shown in Fig. 2B, the migration rate of EPCs gradually rose with increasing concentrations of CXCL12, with an about 3-fold increase over control at 80 ng/mL, suggesting that CXCL12 enhanced EPC migration in a dose-dependent manner.
To address the question of whether CXCL12 could have an effect on EPC adhesion, cells were stimulated with various doses of CXCL12 followed by a cell-adhesion assay. As shown in Fig. 2C, preconditioning with 20 ng/mL CXCL12 significantly increased the number of adherent cells, with an about 2-fold increase in the 80 ng/mL CXCL12-treated groups. These results showed that CXCL12 dose-dependently induced EPC adhesion.
CXCR4 was responsible for CXCL12-induced EPC proliferation, migration, and adhesion
Reports show that increased surface CXCR4 expression was associated with enhanced EPC migration (Chen and others 2010). To determine whether CXCL12 enhanced EPC proliferation, migration, and adhesion via CXCR4 pathway, CXCR4 inhibitor AMD3100 was applied in the medium for 24 h. Flow cytometry analysis of surface CXCR4 expression and western blotting analysis showed that CXCL12 tended to increase surface CXCR4 expression compared with control (Fig. 3A, B).

CXCL12 induced EPC proliferation, migration, and adhesion though CXCR4 signaling. EPCs were stimulated with 80 ng/mL CXCL12, then the protein levels of CXCR4 were examined by western blotting
Further analysis of EPC proliferation, migration, and adhesion showed that compared with the control group, AMD3100 pretreatment significantly restrained the increase in proliferation (Fig. 3C) migration (Fig. 3D), and adhesion (Fig. 3E) induced by CXCL12. All of these results revealed that CXCL12 extracellularly triggered the proliferation, migration, and adhesion of EPCs, through CXCR4 signaling pathways.
Effect of CXCL12 on EPC differentiation into ECs in vitro
EPCs have the potential to differentiate into ECs, which are necessary for angiogenesis. vWF, Tie-2, and VE-cadherin are known to be common markers of ECs. To explore whether CXCL12 could induce EPCs differentiated into ECs in vitro, RT-PCR and western blotting were performed. After stimulation with CXCL12 for 14 days, vWF mRNA was significantly increased, and similar increases in mRNA levels of Tie-2 and VE-cadherin were also confirmed (Fig. 4A). While protein levels showed a similar trend in treatment groups compared with the control group (Fig. 4B), further analysis of the mechanism confirmed that pretreatment with AMD3100 led to an obvious decrease in the mRNA and protein levels of the 3 molecules. All these results suggested that CXCL12 induced EPC differentiation into ECs through CXCR4 signaling pathways.
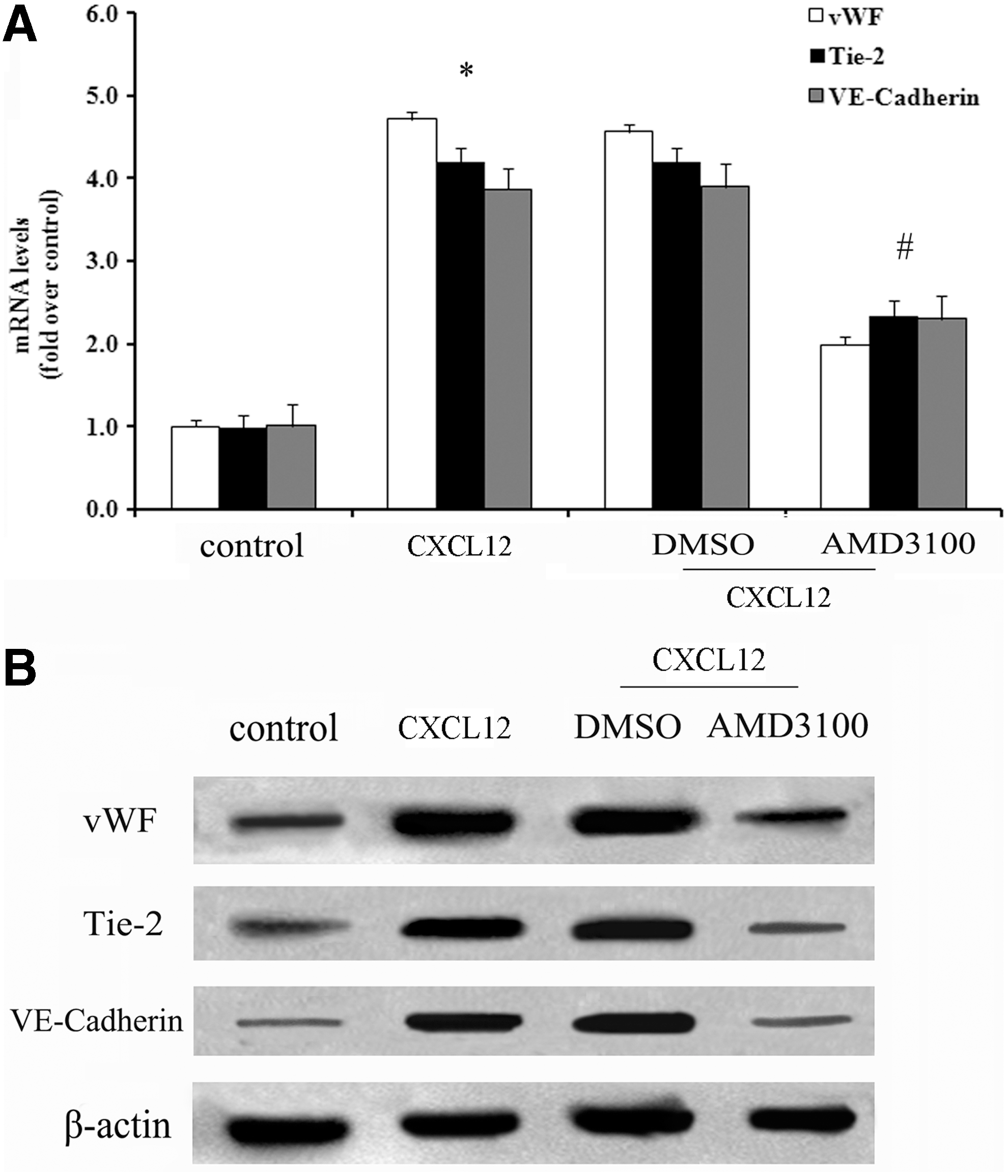
CXCL12 triggered the differentiation of EPCs into endothelial cells. Following preconditioning with CXCR4 inhibitor AMD3100, or not, cells were stimulated with 80 ng/mL CXCL12. Then, the mRNA levels of the endothelial markers vWF, Tie-2, and VE-cadherin were analyzed by real-time polymerase chain reaction, and the corresponding quantitative analysis was performed
CXCL12 induces angiogenesis by CXCR4 signaling in vitro and in vivo
To investigate whether CXCL12 could stimulate angiogenesis, a tube formation assay was utilized. As shown in Fig. 5A, CXCL12 stimulated well-organized, capillary-like networks that were not present in the control group. These results indicated that CXCL12 had angiogenic properties in vitro. Subsequently, we evaluated the angiogenic activity of CXCL12 in vivo by a Matrigel plug assay. Higher fluorescent signals for CD31, an endothelial marker, were measured by immunostaining compared with the control group, suggesting that CXCL12 stimulated blood vessel formation (Fig. 5B). Then, we silenced CXCR4 expression in EPCs by AMD3100 to determine whether the pro-angiogenic activity of CXCL12 was correlated with CXCR4. As expected, disruption of CXCR4 signaling blocked CXCL12-mediated organization of capillary networks (Fig. 5C). Taken together, these results indicated that CXCL12 could act as an angiogenic factor in vitro and in vivo to induce angiogenesis, through the CXCR4 pathway.
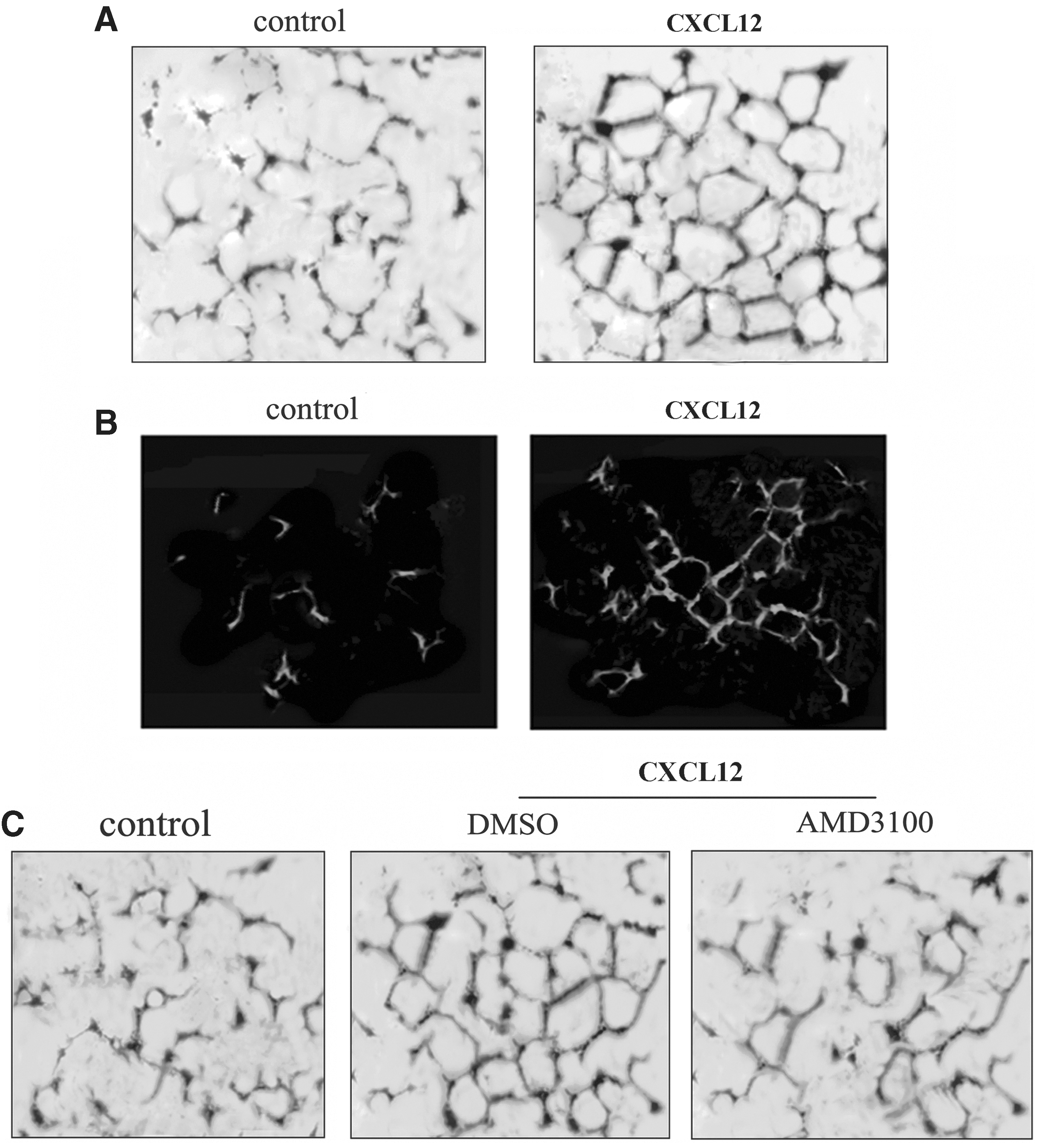
CXCL12 stimulated angiogenesis in vitro and in vivo. After incubation for 20 h in M199 medium containing the indicated test compounds, tube formation was monitored by Diff-Quik® staining
Angiogenesis in a rat ICH model
Based on the previous results, we then assessed the possible activity of CXCL12/CXCR4 in angiogenesis in vivo. After constructing a rat model of ICH, 4 experimental groups were treated respectively as described. After 14 days, the rats' brains were cut into slices. Immunohistochemistry was used to observe EC marker vWF. The results indicated that EPCs could promote angiogenesis compared with the control (G2 vs. G1); when delivered together with CXCL12, higher blood vessel formation was observed (G3). However, this effect was impeded by injection of AMD3100 (G4) (Fig. 6A), the number of blood vessel formation was also shown in Fig. 6B. Thus, these data confirmed the pro-angiogenesis activity of CXCL12 after ICH in vivo, through the CXCR4 pathway.
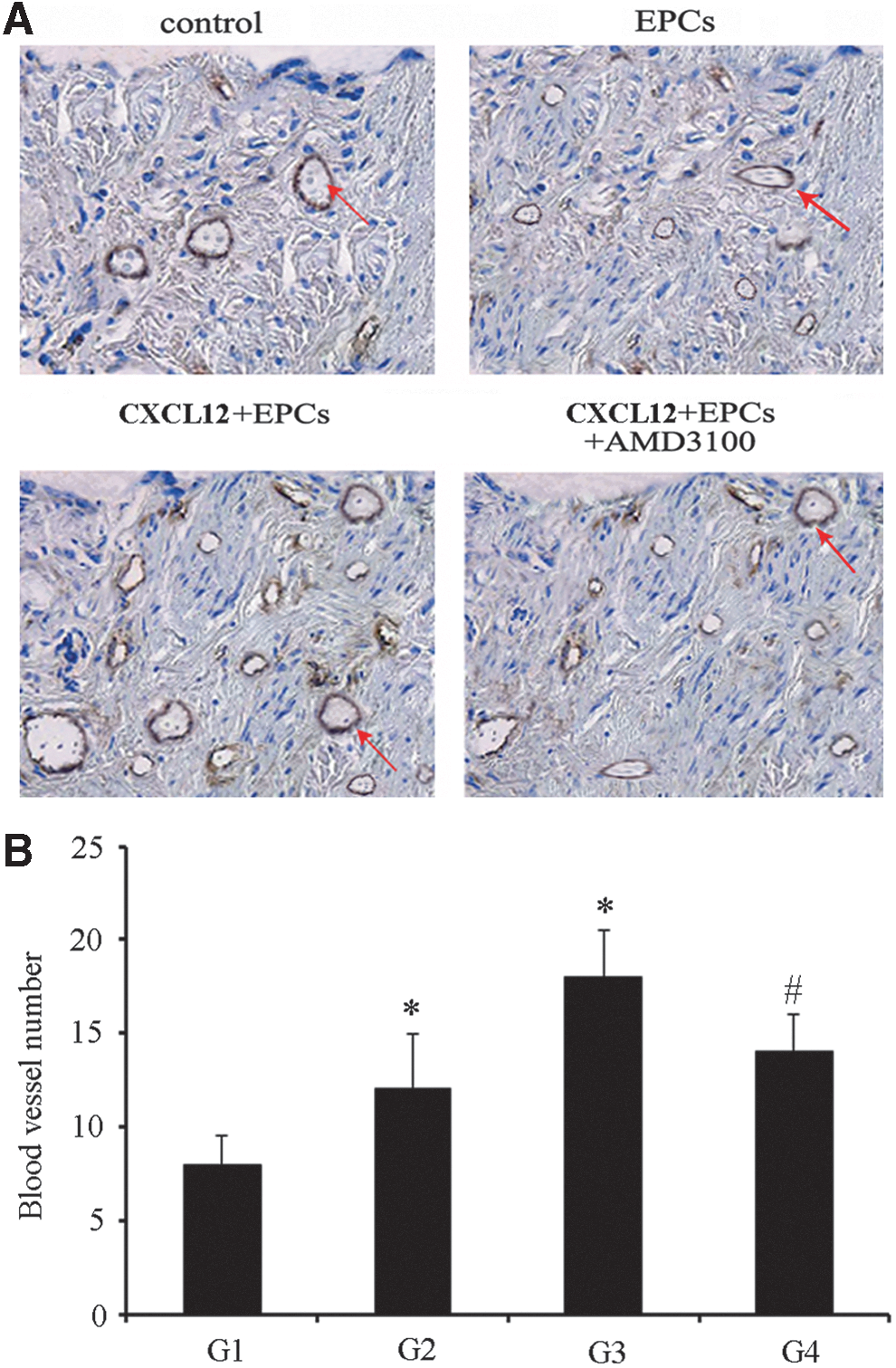
CXCL12 induced angiogenesis in a rat ICH model. Following construction of a rat ICH model, EPCs and/or CXCL12 were injected into the tail vein, with or without subsequent injections of AMD3100. Two weeks later, vWF staining was used to label the new vessels (red arrows)
Discussion
ICH is an often fatal type of stroke. If the patient survives after ictus, the resulting hematoma within the brain parenchyma triggers a series of adverse events, causing secondary insults and severe neurological deficits (Lauritzen and others 2010). Early completion of vascular repair and angiogenesis may effectively reduce secondary injury (Aronowski and Zhao 2011). EPCs, also known as vascular endothelial stem cells, are a kind of precursor cell with high proliferative potential, which can differentiate into mature ECs under certain conditions (Timmermans and others 2009). Studies have shown that EPCs play an important role in ischemia and vascular injury, both in physiology and pathophysiology after birth (Fan and others 2010; Caballero and others 2013; Li and others 2013). When vascular injury happened, EPCs from the bone marrow can be mobilized in the peripheral circulation by outward migration and homing to sites of vascular injury or ischemia, and then differentiate into mature ECs, to promote the formation of new vessels to repair damaged vessels and relieve symptoms of tissue defects (Briasoulis and others 2011). Accordingly, the molecules involved in angiogenesis and osteogenesis have a potential clinical prospect in ICH therapy.
Chemokines play an important role in many functions of stem cells. A study has found that when tissue damage or inflammation occurs, a large number of cytokines are released into the blood and take part in tissue repair (Ziebell and Morganti-Kossmann 2010). CXCL12 is a member of the CXC subfamily. It can activate a variety of stem cell chemoattractant properties and is always highly expressed in the damaged area after injury (Zlotnik and Yoshie 2000). The upregulation of CXCL12 also plays an important role in the mobilization of EPCs from the bone marrow into blood (Carr and others 2006). Further research shows that CXCL12 and its receptor CXCR4 are important in stem cell migration and homing behavior (Miller and others 2008). However, it was unclear whether CXCL12 could induce EPCs to regulate angiogenesis and osteogenesis. In this study, CXCL12 dose-dependently induced the proliferation, migration, and adhesion of EPCs. Analysis of the mechanism showed that CXCR4 inhibitor AMD3100 pretreatment could significantly restrain the increase of proliferation, migration, and adhesion induced by CXCL12. The results revealed that CXCL12 extracellularly triggered the proliferation, migration, and adhesion of EPCs, through CXCR4 signaling pathways, which suggests a potential angiogenic activity of CXCL12.
Proliferation and differentiation of EPCs are closely related to angiogenesis (Palladino and others 2012). To clarify whether CXCL12/CXCR4 plays roles in angiogenesis in this experiment, we then investigated the expression of EC markers in vitro. The differentiation of EPCs was assessed by detecting the EC markers vWF, Tie-2, and VE-cadherin. As expected, CXCL12 can upregulate the expression of endothelial marker vWF, Tie-2, and VE-cadherin, which was blocked by AMD3100 pretreatment. Therefore, these results confirmed that CXCL12 could induce EPCs to differentiate into ECs, through CXCR4 signaling pathways. Tube formation has an important part in angiogenesis; studies have shown that by inducing apoptosis in tube-forming ECs can effectively suppress angiogenesis (Kunimasa and others 2011). Further analysis suggested that a notable increase in the formation of capillary tubes was observed after CXCL12 preconditioning. CD31 is a known marker of blood vessels and its strong immunofluorescence signals were also confirmed in vivo by a Matrigel plug assay, suggesting a potential pro-angiogenic activity of CXCL12.
During ICH, rapid accumulation of blood within the brain parenchyma leads to disruption of normal anatomy and increased local pressure (Li 2012). Vascular repair and angiogenesis play an important role in ICH-induced brain injury (Lei and others 2013). To further confirm the function of CXCL12 in angiogenesis after ICH, we constructed a rodent model of ICH. As expected, injection of CXCL12 and EPCs obviously increased newborn vascular numbers at the damaged brain regions compared with the control and EPC groups. Interestingly, AMD3100 delivery significantly attenuated the effect of CXCL12 on angiogenesis. Hence, our data have confirmed that application of exogenous CXCL12 with EPCs can stimulate angiogenesis through the CXCR4 pathway.
In this study, CXCL12 dose-dependently enhanced EPC proliferation, migration, and adhesion, stimulated angiogenesis via activation of the CXCR4 pathway, and thereby induced angiogenesis after ICH. These findings identify the potential pro-angiogenic activity of CXCL12 after ICH. Thus, our study may provide a new therapeutic strategy for ICH. When ICH happened, in the early stage timely application of CXCL12 and EPCs may promote early vascular repair and angiogenesis, improving the environment of damaged regions, and they play an important role in prevention of secondary neuronal injury and subsequent neuron regeneration.
Footnotes
Author Disclosure Statement
No competing financial interests exist.