
Abstract
Human immunodeficiency virus-1 (HIV-1)-infected monocytes/macrophages and microglia release increased levels of proinflammatory cytokines and chemokines, including ELR+ (containing glutamic acid–leucine–arginine motif) chemokines. To investigate the role of HIV-1 infection on chemokine regulation, monocyte-derived macrophages (MDMs) from normal donors were infected with HIV-1 and the expression of chemokines and their downstream biological functions were evaluated. Among the tested chemokines, CXCL5 was upregulated significantly both at the mRNA and protein level in the HIV-1-infected MDMs compared with mock-infected cultures. Upregulation of CXCL5 in the HIV-1-infected MDMs is, in part, regulated by increased interleukin-1β (IL-1β) production and phosphorylation of ERK1/2. Functional analyses indicate that HIV-1-induced overexpression of CXCL5 has enhanced the ability to attract neutrophils, as observed by chemotaxis assay. However, exposure of NT2, SH-SY5Y cells, and primary neurons to HIV-1-infected MDM supernatants resulted in cell death that was not rescued by anti-CXCL5 antibody suggesting that CXCL5 does not have direct effect on neuronal death. Together, these results suggest that the increased level of CXCL5 in tissue compartments, including the central nervous system of HIV-1-infected individuals might alter the inflammatory response through the infiltration of neutrophils into tissue compartment, thus causing secondary effects on resident cells.
Introduction
H
HIV-1 invades the CNS during the early phase of infection through migrating perivascular monocytes/macrophages and lymphocytes (Ho and others 1985; Palmer and others 1994; Lipton and Gendelman 1995; Zink and others 1999). Studies indicate that 30%–60% of HIV-1-positive patients develop mild to severe forms of neurological disorders even in the absence of detectable virus in the periphery due to anti-retroviral therapy (ART) (Skinner and others 2009; Becker and others 2013; Gelman and others 2013). HIV-1-induced neuropathogenesis is, in part, mediated by proinflammatory cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and chemokines (Merrill and others 1989; Tyor and others 1992; Brabers and Nottet 2006; Gandhi and others 2009). HIV-1 viral proteins gp120, Tat, and Vpr stimulate IL-1β expression in monocytes/macrophages (Cheung and others 2008; Yang and others 2010; Guha and others 2012) suggesting that HIV-1 exerts inflammatory effects in CNS through both direct and indirect methods. There is also evidence that IL-1β has a potent role in stimulating chemokines, including CXCL5, CCL2, CCL5, and CXCL1 (Unemori and others 1993).
Expression of proinflammatory cytokines and chemokines are induced by HIV-1 exposure and infection (Katsikis and others 2011). This upregulation of cytokines and chemokines by the virus and viral proteins are mediated by signaling molecules, including mitogen-activated protein kinase (MAPK). HIV-1-induced phosphorylation of ERK1/2 and p38 (Wilflingseder and others 2004; Guha and others 2012; Crawford and others 2013) increases the expression of cytokines and chemokines in multiple cell types (D'Aversa and others 2004; Williams and others 2009; Furler and Uittenbogaart 2010). Binding of gp120 with the chemokine receptors activates c-Jun amino-terminal kinase/stress-activated protein kinase and p38. Furthermore, activation of macrophages by gp120 resulted in the increased expression of chemokines CCL2 and MIP-1β through MAPK activation (Del Corno and others 2001). Similarly, HIV-1 protein Nef stimulates the production of CCL5 in astrocytes by p38/MAPK and PI3K/Akt pathway (Liu and others 2014).
In this study, we investigated the role of HIV-1 infection on dysregulation of chemokine expression, their transcriptional regulation, and its consequences in chemotaxis and neuronal survival. Results indicate that HIV-1 increases the CXCL5 expression significantly both at the mRNA and protein levels in HIV-1-infected monocyte-derived macrophages (MDMs) compared with mock-infected cultures. Upregulation of CXCL5 in HIV-1-infected MDMs was, in part, regulated by increased IL-1β and increased ERK1/2 phosphorylation. Enhanced secretion of CXCL5 by HIV-1-infected MDMs did not have any direct effect on neuronal viability; however, it enhanced the neutrophil infiltration as observed in the chemotaxis assay. Together, these results suggest that HIV-1 infection in macrophages/microglia dysregulates chemokines expression through IL-1β and phosphorylation of MAPK kinases that results in the infiltration of neutrophil into tissue compartments.
Materials and Methods
Reagents
HIV-1 YU2 plasmid was obtained from Dr. Serge Benichou, France. CXCL5 promoter construct was a kind gift from Prof. Lluis Fajas Coll, Switzerland. Neural progenitor (NP) cells were purchased from Millipore, and human recombinant IL-1β and neutralizing antibody against IL-1β were purchased from R&D Systems. Recombinant CXCL5 was from Peprotech and anti-CXCL5 was purchased from Abcam. ERK1/2, p38, and SAPK/JNK inhibitors (PD98059, SB203580, and SP600125 respectively) were purchased from Calbiochem.
Isolation and culture of MDMs
MDMs were generated from normal peripheral blood mononuclear cells (PBMC). Heparinized blood samples were purchased from the Pittsburgh Blood Bank using appropriate IRB approvals from the University of Pittsburgh. Briefly, PBMCs were isolated by Ficoll-Hypaque gradient centrifugation. CD14+ monocytes were purified by positive selection using anti-CD14 monoclonal antibody-coated magnetic microbeads (Miltenyi Biotech) and cultured as described previously (Schafer and others 2006).
Generation of CXCR2+cells and isolation of neutrophils for chemotaxis
Cells stably expressing human CXCR2 were generated by transfection and selection as described (Qin and others 2008). The human CXCR2 cDNA clone was provided by the Missouri S&T cDNA Resource Center. Single cell clones were subjected to 2 rounds of single cell cloning and screened for high-dose responsiveness to CXCL5 (Peprotech). Neutrophils were isolated from whole blood by density gradient method as described previously (Freitas and others 2008). Briefly, 3 mL of Histopaque 1077 was layered on 3 mL of Histopaque 1119, and 6 mL blood was layered on top of the Histopaque layers, and the tube was centrifuged at 890 g for 30 min at 20°C. Neutrophils were removed carefully and the volume was doubled with phosphate buffered saline (PBS). The residual red blood cells (RBC) were lysed by RBC lysis solution. The isolated neutrophils were used immediately.
Culture of neuronal cell lines and primary neurons
NT2 (Human teratocarcinoma cell line), SH-SY5Y (human neuroblastoma cell line), and HeLa cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. NP cells at passage 2–6 were maintained in 35-mm plates coated with 20 μg/mL poly-
Virus preparation and treatment of macrophages
HEK293T cells were transfected with 5 μg of HIV-1 proviral construct, HIV-1 YU2, or vector DNA (for mock) using the PolyJet Reagent (SignaGen Laboratories) as per the manufacturer's instructions. Seventy-two hours posttransfection, supernatants were collected, spun at 3,000 rpm for 10 min, and filtered through a 0.22 μm filter to remove cellular debris. Virus titer was measured by p24 enzyme-linked immunosorbent assay (ELISA) and infectivity of the viruses was calculated by standard TZM-bl assay. Differentiated (7–8 day old) MDMs were either infected with HIV-1 at a multiplicity of infection (MOI) of 0.1 or exposed to vector transfected supernatant from HEK293T culture as negative control for mock infection. HIV-1-infected and -control MDMs were maintained for 21 days. Cell pellets and supernatants were collected every 24 h up to day 4 (exposure effect) and at 4 days interval from day 8 to 20 to monitor virus replication and chemokine production. For assessing chemokines in the presence of MAPK inhibitors, MDMs were pretreated with 10 μM of PD98059 (ERK1/2 inhibitor), SB203580 (p38 inhibitor), or SP600125 (SAPK/JNK inhibitor) for 2 h followed by infection with HIV-1 at an MOI of 0.1 or mock. DMSO was used as vehicle control.
Proinflammatory chemokine array profiling by quantitative real-time PCR
At multiple time points postinfection, MDMs were washed with cold PBS and total RNA was extracted using the RNeasy mini kit (Qiagen) according to the manufacturer's protocol, with additional on-column DNase1 digestion (RNase-free DNase kit; Qiagen). The RT2 Profiler PCR Array (SABiosciences) was used for mRNA profiling studies and the assay was performed according to the manufacturer's protocol. Briefly, 1 μg of total RNA extracted from mock- or HIV-1-infected MDMs was converted to cDNA using the RNA first-strand synthesis kit. The cDNA product was used to perform gene expression array using a Taqman 7900HT machine. Data were normalized with endogenous controls (included in the array) and differential regulation of proinflammatory chemokines (fold change) was analyzed from the Ct values from day 0 to 20 using the SABiosciences web-based tools. Genes that are differentially regulated (±2-fold) in HIV-1-infected cultures were determined.
Measurement of CXCL5 and IL-1β by ELISA
Amount of CXCL5 and IL-1β released into the supernatant collected from HIV-1- or mock-infected MDMs at specific time points were measured by ELISA as per the manufacturer's protocol (R&D Systems). The optical density (OD) was determined using the ELISA plate reader and the concentration of the CXCL5 and IL-1β were calculated from the standard curve. For blocking experiments, MDMs were treated with 5 μg/mL neutralizing antibody against IL-1β or IgG control antibody for 2 h before HIV-1 infection. Supernatants were harvested at 3, 6, 12, and 24 h postinfection and the level of CXCL5 was measured.
Luciferase assay
HEK293T and HeLa cells (0.25×106 cells/well) in a 24-well plate were transfected with 1 μg of the reporter plasmid containing CXCL5 promoter-driven luciferase gene using PolyJet (SingaGen). Posttransfection (36 h) cells were treated with either 1, 10, and 20 ng of recombinant human IL-1β (rhIL-1β) or MDM supernatants from HIV-1- or mock-infected cultures. Reporter activity was measured 3 h posttreatment as suggested by the manufacturer (Promega) and results are represented as relative light unit.
Treatment of neuronal cell lines and primary neurons and apoptosis analyses
Briefly, primary neurons (3×104 cells/well) as well as NT2 and SH-SY5Y cells were exposed to HIV-1- or mock-infected MDM supernatants (50% of the volume of total media) in the presence or absence of a neutralizing antibody against CXCL5 (10 μg/mL). To detect neuronal apoptosis, cells were washed twice with 1×PBS and were stained with the JC-1 staining solution for 15 min at 37°C. Healthy cells were detected at Texas Red filter and apoptotic cells were detected using FITC channel. Apoptosis (%) was quantified by nuclei staining with Hoechst 33342 and neuronal apoptosis was calculated from the percentage of cells stained with JC-1.
Chemotaxis assay
Chemotaxis using CXCR2+ L1.2 cells or neutrophils was performed as described (Qin and others 2008) using 96-well chemotaxis plates (Neuro Probe) in 3 independent experiments, each performed with triplicate wells. Briefly, 28 μL of control or HIV-1-infected MDM supernatants were added in the bottom well, filters with 5 μM pore size were placed between bottom plates and top plates of the chamber and the neutrophils or CXCR2-expressing cells were added to the top wells. The chamber was incubated for 2 h at 37°C and 5% CO2. The number of migrated cells was counted under microscope. Multiple concentrations (1, 10, and 100 nM) of rhCXCL5 were used as positive control. For blocking experiments, MDMs were pretreated with 5 μg of anti-CXCL5 for 2 h followed by HIV-1 or mock infection and anti-CXCL5 was maintained during the infection period. Supernatants were harvested at 48–72 h postinfection and used for chemotaxis assay to monitor the migration of neutrophils. Fold reduction was calculated using MDM supernatants with isotype control (IgG control) as negative control.
Western blot
MDMs (HIV-1/mock or infected) were washed twice with cold PBS and lysed in RIPA buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 1.0 mM phenylmethyl-sulfonylfluoride, 0.05% deoxycholate, 10% SDS, 0.07 trypsin inhibitor units/mL aprotinin, and protease inhibitors Leupeptin, Chymostatin, and Pepstatin (1 μg/mL). Cell lysates were clarified by centrifugation and total cell lysates (50 μg equivalent) were separated on a SDS-PAGE gel, transferred, and protein expression was detected with anti-ERK1/2 (total and active; Cell Signaling Technology), anti-p38 (total and active; Cell Signaling Technology,), and anti-SAPK/JNK (active) (Cell Signaling Technology). β-actin (Cell Signaling Technology) was used as loading control. Blots were developed using the ECL kit (Pierce). The intensity of the bands was quantitated by the ImageJ software and the densitometric measurements were normalized against the total protein expression levels for MAPKs.
Statistical analysis
Results were expressed as mean+ SEM for 3 or more independent experiments. The data were analyzed using Student's t-test for normally distributed data with equal variances and P≤0.05 was considered as significant. The immunoblotting images were quantified using the ImageJ software. For quantitative real-time PCR (qRT-PCR) data analyses the SABiosciences web-based software was used.
Results
HIV-1 induced differential regulation of chemokines in MDMs
Proinflammatory cytokines and chemokines released by macrophages and microglia in the CNS compartment play a crucial role in HIV-1-induced neuropathogenesis (Klein and others 1999; Buch and others 2004; El-Hage and others 2008). To examine the effect of HIV-1 on chemokine regulation, normal donor-derived MDMs were infected with HIV-1 at an MOI of 0.1 or mock infected and were assessed for a panel of chemokines by qRT-PCR array. Compared with mock-infected controls, HIV-1-infected MDMs differentially expressed a number of chemokines at different time points postinfection (Table 1). Among the upregulated genes, CXCL5 and CXCL3 showed consistent increase over mock infected throughout the tested time points (fold change ranged from 2 to >500 for CXCL5 and 2 to >40 for CXCL3), whereas, CCL23, CXCL6, and CXCL14 showed a modest increase with varying expression levels during early (exposure) and late (replicative) phases of infection. During the replication phase (8–20 days postinfection) CXCL2, CXCL5, and CXCL3 remained at high levels, whereas the other chemokines were either not altered or did not show any consistent dysregulation in HIV-1-infected MDMs compared with mock-infected MDMs. Furthermore, combined analyses, including multiple time points postinfection singled out CXCL5 as the most significant chemokine exhibiting 57.6-fold increase over control with the significance of P=0.022. Therefore, we focused the rest of this study to delineate the expression pattern of CXCL5, its regulation and its functional consequences in HIV-1 pathogenesis.
HIV-1 infection dysregulates inflammatory chemokine genes in MDMs. HIV-1-infected and mock-infected MDMs were assessed by qRT-PCR for the expression of inflammatory chemokines up to day 20 postinfection and fold changes in HIV-1-infected MDMs were calculated compared with mock-infected MDMs. Genes with more than 2-fold change were only considered. Positive values indicate upregulation and negative values downregulation of inflammatory chemokine genes in HIV-1-infected MDMs compared with mock.
qRT-PCR, quantitative real-time PCR.
To further confirm whether the differences noted in HIV-1-induced CXCL5 production in MDMs at the transcriptional level is reflected at the protein level as well, in supernatants from HIV-1- and mock-infected MDMs CXCL5 level was measured by ELISA. Consistent with the changes in mRNA, the concentration of secreted CXCL5 increased drastically in HIV-1-infected MDMs compared with mock-infected throughout the course of infection in multiple donors (Fig. 1A). Depending on the donors, the fold change ranged from 3- to 9-fold higher in the infected culture supernatants compared with the mock-infected culture supernatants. Combined analysis (n=6) showed significant increase of CXCL5 in HIV-1-infected MDMs over mock-infected MDMs at different time points postinfection (Fig. 1B). Together, the results indicate that HIV-1 infection increases the level of CXCL5 both at the mRNA and protein level in MDMs.
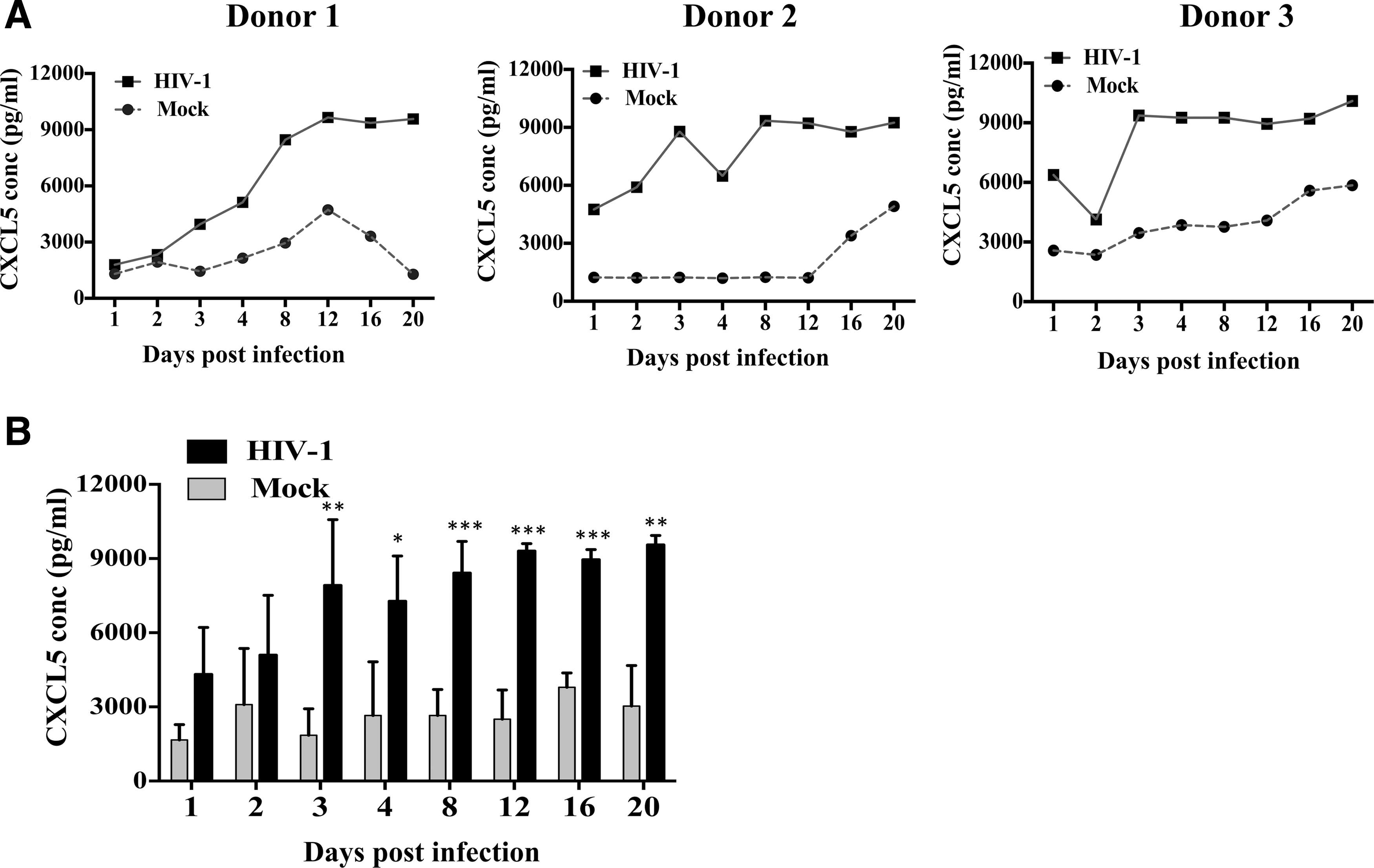
HIV-1 infection enhances expression of CXCL5 in monocyte-derived macrophages (MDMs). MDMs were infected with HIV-1 at an MOI of 0.1 or mock. Cells and supernatants were harvested at 1, 2, 3, 4, 8, 12, 16, and 20 days postinfection.
HIV-1 regulates the expression of CXCL5 through IL-1β in MDMs
Previous studies have shown that proinflammatory cytokine, IL-1β triggers the expression of CXCL5 in different cell types (Bersinger and others 2011; Carrero and others 2012; Okabe and others 2012). To understand whether HIV-1 utilizes similar mechanisms to upregulate CXCL5 expression in infected MDMs, first, we compared the level of IL-1β and CXCL5 at the mRNA and protein level in the same samples. Results from the mRNA transcript analyses indicate that IL-1β and CXCL5 have parallel expression pattern and infection/exposure of HIV-1 induced similar IL-1β and CXCL5 dysregulation in MDMs (Fig. 2A). The change in mRNA level was further reflected in both IL-1β and CXCL5 at the protein level as expected (Fig. 2B).
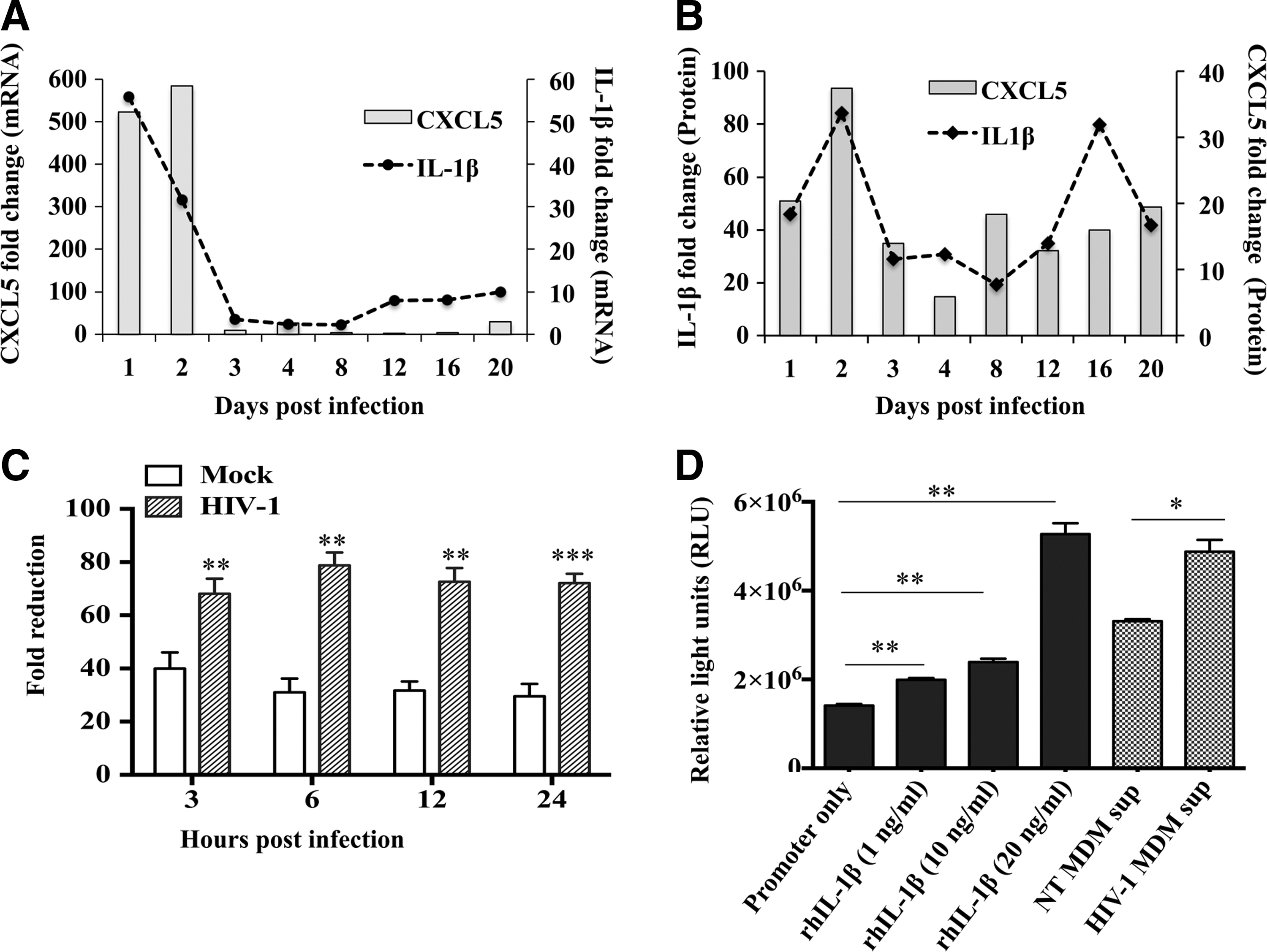
Expression of CXCL5 is induced, in part, by IL-1β. MDMs were infected with either HIV-1 at an MOI of 0.1 or mock. Cells and supernatants were harvested at 1, 2, 3, 4, 8, 12, 16, and 20 days postinfection.
To determine whether IL-1β directly modulates CXCL5 production, MDMs were pretreated with anti-IL-1β antibody or IgG control antibody before HIV-1 infection. Supernatants from HIV-1- and mock-infected MDMs were collected 3–24 h postinfections and CXCL5 level was measured. Fold changes were calculated using HIV-1- and mock-infected MDM supernatants with IgG control antibody as control. CXCL5 expression data in both NT and HIV-1-infected supernatants post-anti-IL-1β antibody treatment were normalized over that of IgG control. Results indicate that there is a significant (P<0.05) reduction in CXCL5 level in HIV-1-infected MDM supernatants compared with mock-infected cultures at 3, 6, 12, and 24 h postinfection (Fig. 2C). To further understand whether IL-1β activates CXCL5 expression through IL-1β response elements in CXCL5 promoter, we transfected HEK293T cells with a reporter plasmid containing CXCL5 promoter-driven luciferase gene and treated with different amounts of rhIL-1β. Reporter activity measured posttreatment exhibited a dose-dependent increase in CXCL5 promoter activity by rhIL-1β stimulation compared with no stimulation (Fig. 2D). The fold increase was 1.4, 1.7, and 3.7 for 1, 10, and 20 ng of IL-1β treatment with the significance of 0.0042, 0.0037, and 0.0021, respectively. Additionally, a significantly (P=0.0145) higher level of luciferase gene expression was also observed in these cells treated with HIV-1-infected MDM supernatants compared with mock-infected supernatants, further confirming that IL-1β is, in part, responsible for increased CXCL5 expression. To examine if the effect of IL-1β on CXCL5 promoter is cell specific, the experiment was repeated with HeLa cells and similar results were observed (data not shown).
Regulation of CXCL5 by MAPK signaling in HIV-1-infected MDMs
In addition to transcriptional regulation by IL-1β, expression of chemokines, including ELR+ chemokines are regulated by MAPK signaling (Kojima and others 2010; Baston-Bust and others 2013). Therefore, next, we examined whether MAPK family members, ERK1/2, p38, and SAPK/JNK have any role in CXCL5 production in MDMs. First, phosphorylation of ERK1/2, p38, and SAPK/JNK was assessed in HIV-1-exposed/infected and control MDM cell lysates and the bands were quantitated by densitometry (Fig. 3A, B). Results indicate a significant increase in phosphorylation of ERK1/2 and p38 within 30 min postexposure in HIV-1-treated MDMs compared with mock-treated culture. However, SAPK/JNK showed no significant difference between these groups (data not shown) indicating the specificity and involvement of ERK1/2 and p38 pathways in HIV-1 infection.
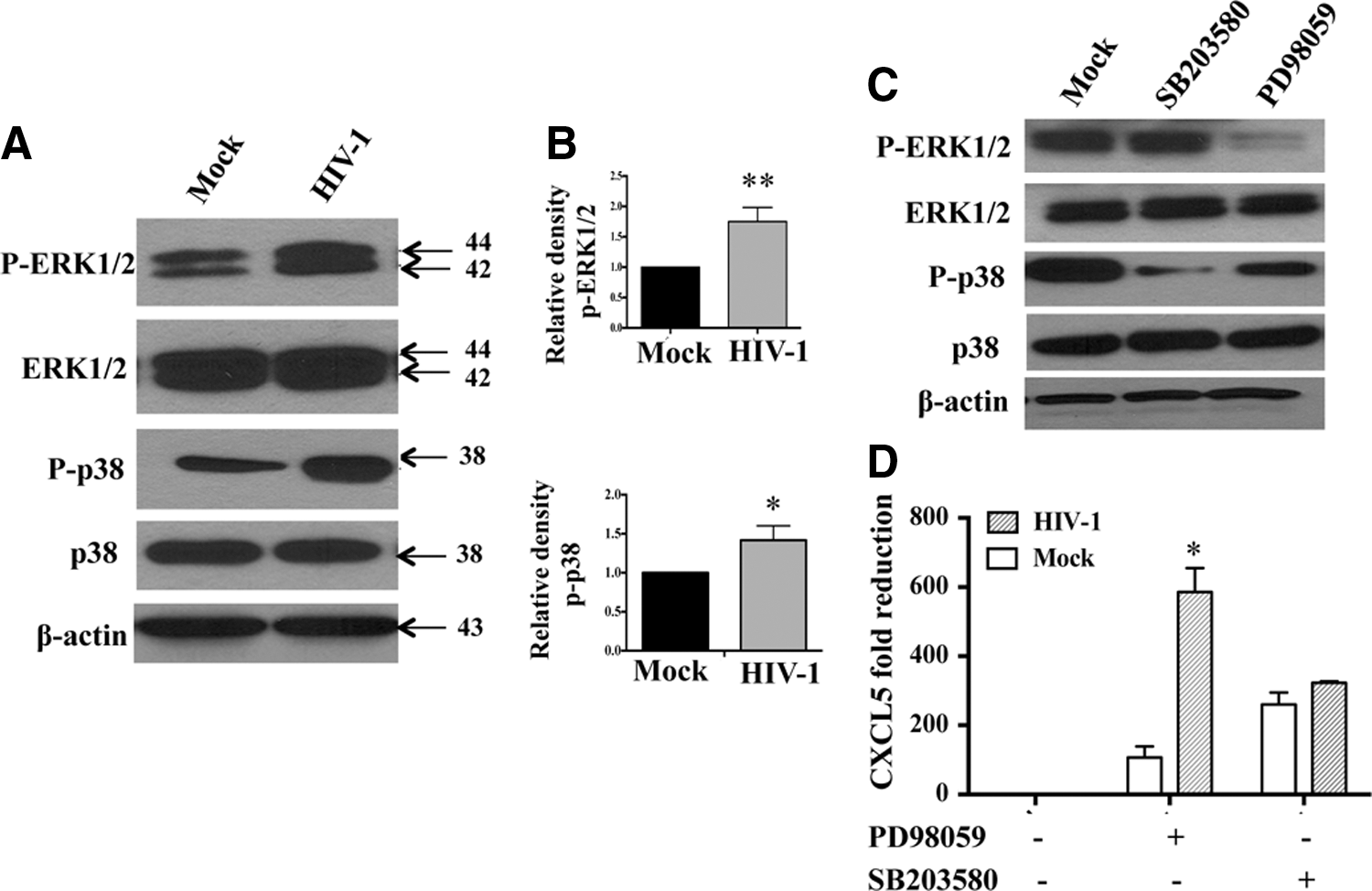
HIV-1-induced CXCL5 production is regulated by ERK1/2 signaling in MDMs. MDMs were infected with HIV-1 at an MOI of 0.1 or mock and the cells were harvested 30 min posttreatment.
Next, to assess whether increased phosphorylation of ERK1/2 and p38 is associated with the upregulation of CXCL5 in HIV-1-infected MDMs, MDMs were treated with SB203580 (p38 inhibitor), PD98059 (ERK1/2 inhibitor), and SP600125 (SAPK/JNK inhibitor) before infection with HIV-1. Figure 3C demonstrates the reduced phosphorylation of ERK1/2 and p38 in HIV-1-infected MDMs compared with control in the presence of specific MAPK inhibitors. Next, CXCL5 was measured in the presence of MAPK inhibitors. Compared with the untreated control group, MAPK inhibitors reduced the levels of CXCL5 production in HIV-1-infected MDMs (Fig. 3D). Blocking ERK1/2 pathway with PD98059 showed significant reduction (5.5-fold reduction; P=0.0125) in CXCL5 level in HIV-1-infected MDMs compared with mock-infected MDMs. However, SB203580, a potent p38 inhibitor, blocked CXCL5 production irrespective of HIV-1 infection and did not induce any significant difference (1.2-fold) between HIV-1- and mock-infected MDM cultures (P=0.1246) indicating that CXCL5 expression is modulated by ERK1/2 and not p38 signaling. However, reduction of CXCL5 level in mock- and HIV-1-infected MDMs after blocking p38 was significant compared with respective controls (without p38 inhibitor). As expected, SP600125, a selective inhibitor of JNK, also had no significant effect on CXCL5 production in HIV-1-infected or mock-infected MDM culture (data not shown).
Infiltration of neutrophils by CXCL5
One of the major functions of an ELR+ chemokine is to attract granulocytes and the expression of those chemokines parallels with the migration and recruitment of neutrophils into the CNS and other tissue compartments (Hosking and others 2009; Mei and others 2010). To determine whether increased CXCL5 level results in increased recruitment of cells, a chemotaxis assay was performed with a CXCR2 (receptor for CXCL5 ligand)-expressing cell line (L1.2 cells) as well as primary neutrophils using HIV-1- or mock-infected MDM supernatants. Multiple concentrations of recombinant CXCL5 (0, 1, or 10 nM) were used as positive control. Results suggest that CXCL5 recruited neutrophils in a dose-dependent manner (Fig. 4A). At a concentration of 1 nM rhCXCL5, the number of migrated L1.2 cells was 12 times higher compared with mock. However, at 10 nM there was no additional effect on cell migration, suggesting a threshold limit. Similarly, supernatants from HIV-1-infected MDMs induced higher CXCR2 positive cell migration compared with mock-infected MDM supernatants (Fig. 4A). We next sought to investigate whether the upregulation of CXCL5 in HIV-1-infected MDMs had differential impact on the migration of primary neutrophils, which also express the CXCR2 receptor. Similar to L1.2 cells, increased migration of human primary neutrophils was also observed in response to increased concentration of CXCL5 in HIV-1-infected culture supernatants compared with mock-infected supernatants (Fig. 4B). To further confirm the specificity of CXCL5 in neutrophil infiltration, MDMs were preincubated with anti-CXCL5 antibody before HIV-1 or mock infection. Blocking of CXCL5 reduced neutrophil migration by ∼30% in control MDM supernatants whereas, the reduction was ∼65% in HIV-1-positive MDM culture supernatants (P=0.0161) (Fig. 4C). Collectively, these results indicate that CXCL5 plays a major role in the recruitment of neutrophils and cells expressing CXCR2 receptor.
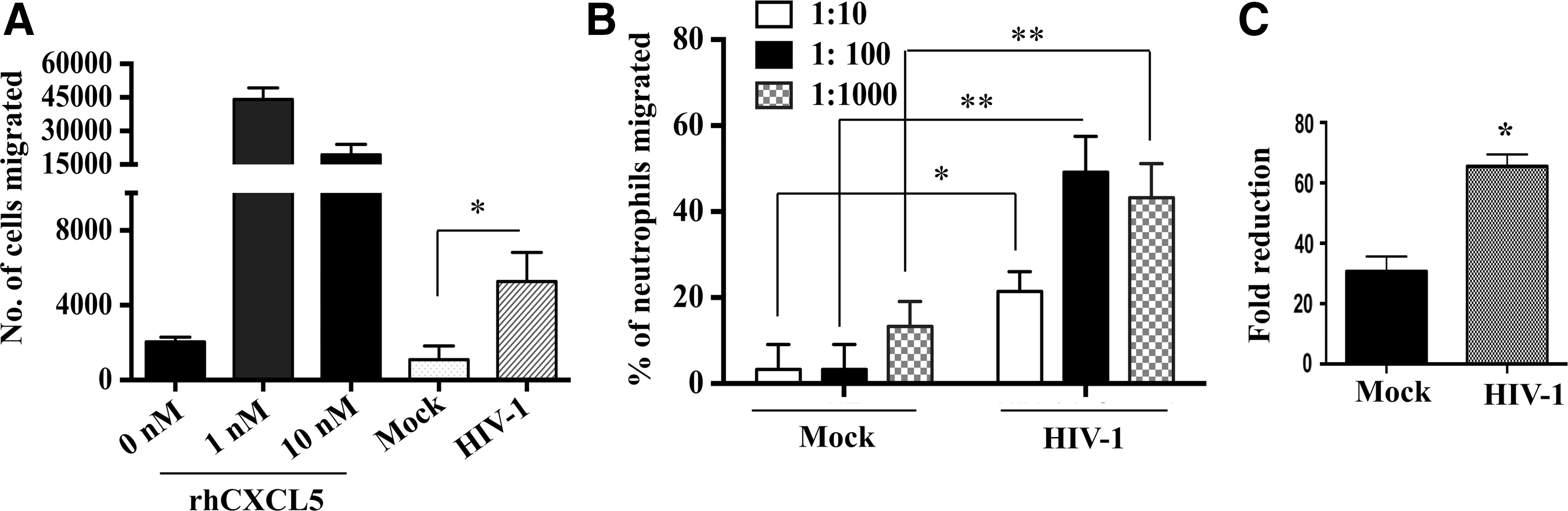
HIV-1-induced CXCL5 enhanced migration of neutrophils and L1.2 cells.
Effect of CXCL5 on neuronal survival
Finally, to investigate the biological consequence of CXCL5 release from macrophages on neurons and CNS pathology, neuronal cell lines NT2, SH-SY5Y, and NP cell-derived human primary neurons were treated with HIV-1- or mock-infected MDM supernatants in the presence or absence of CXCL5 neutralizing antibody or IgG control and apoptosis was measured (Fig. 5A–C). Results indicate that cell death in neuronal cells exposed to control MDM supernatant ranged between 8% to 13% in NT2 and SH-SY5Y cells and 17% in primary neurons, whereas, the cell death increased to 21%–25% in case of NT2 and SH-SY5Y and 37% in primary neurons in case of HIV-1-infected MDM supernatants. Blocking of CXCL5 did not have any significant effect (P>0.05) on death/survival rate in any of these cell types (Fig. 5A–C), suggesting that CXCL5 may not have direct cytotoxic effect on neurons as blocking of CXCL5 did not reduce neuronal survival.
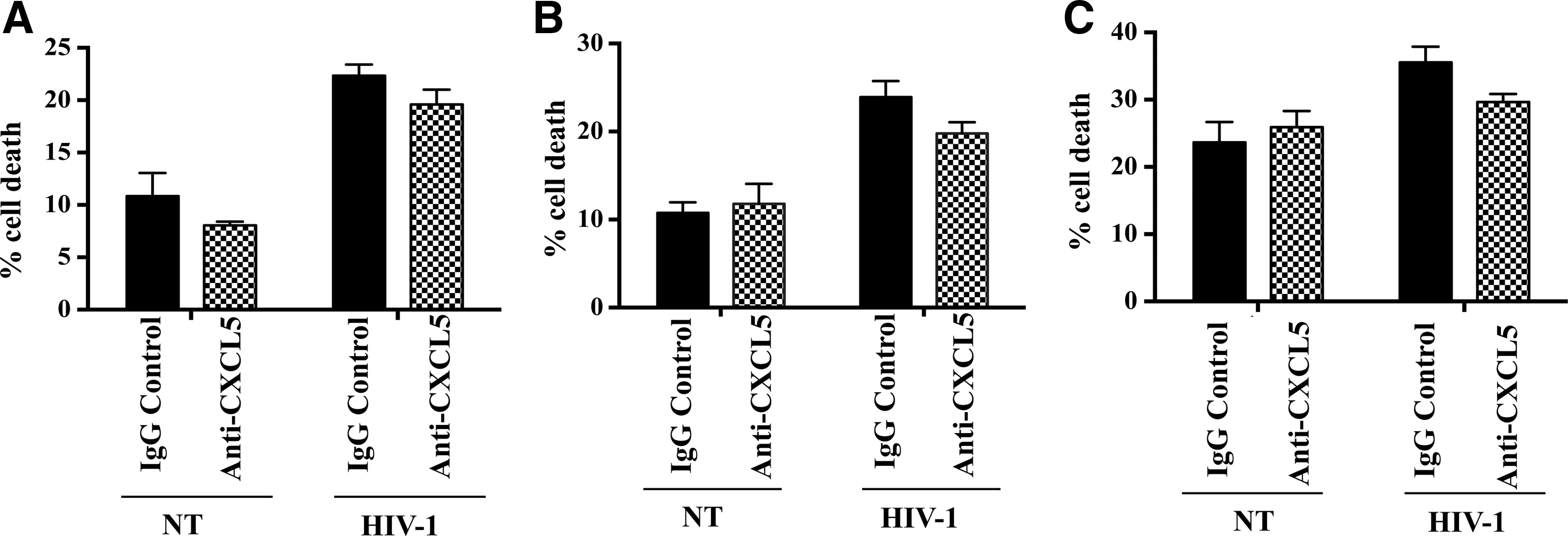
Effect of CXCL5 released by HIV-1-infected MDM on neuronal survival.
Discussion
In this study, we observed dysregulation of chemokines and chemokine receptors in HIV-1-exposed and -infected MDMs. We focused on CXCL5 as it was one of the most significant chemokines dysregulated in MDMs by HIV-1 infection. Previous studies have also shown dysregulation of CXCL5 by HIV-1 infection both in vitro and in vivo (Albright and Gonzalez-Scarano 2004; Duskova and others 2013) as well as by other viruses. For instance, association of CXCL1 and CXCL5 in the West Nile Virus induced encephalitis through IL-22 was reported (Wang and others 2012). Similarly, dysregulation of CXCL5 expression in response to Herpes Simplex Virus (HSV) and Polyomavirus JC (JCV) was observed in CNS compartment (Aravalli and others 2005; Darbinyan and others 2013). However, little is known regarding the regulation of CXCL5 and its functional consequences in the context of HIV-1 infection.
Although the exact mechanism(s) involved in HIV-1-induced CXCL5 upregulation is not fully understood, factors involved at the transcriptional level and signaling pathways are known to modulate the CXCL5 expression in other systems. For instance, IL-1β is known to be regulated by HIV-1 as part of the proinflammatory response both in vivo and in vitro (Devergne and others 1991; Xing and others 2009; Guha and others 2012). It is also a causative factor for severe inflammatory dysfunction in HIV-1-infected subjects (Mamik and others 2011; Ye and others 2013) and induces CXCL5 expression by activating NF-κB and CREB region in CXCL5 promoter (Sun and others 2008). Our results support the notion that IL-1β regulates CXCL5 at the transcriptional level, as we have noted a direct correlation between IL-1β and CXCL5 production in MDMs infected with HIV-1. Reduced production of CXCL5 by blocking IL-1β and direct activation of CXCL5 promoter-driven luciferase gene by IL-1β further confirms the transcriptional regulation of CXCL5. This observation was further supported by computational analysis that demonstrates the presence of seven NF-κB binding sites in the CXCL5 promoter region that could be potentially regulated by IL-1β. Present data further extend our understanding on the functional consequence of the proinflammatory molecule IL-1β on dysregulation of other chemokines, including CXCL5 in context of HIV-1 infection.
Expression of inflammatory cytokines/chemokines and immune activation are hallmarks of HIV-1-associated diseases. Although the number of productively infected cells within HIV-1-infected individuals is very limited, the inflammatory response in infected individuals remains high, suggesting an interaction between HIV-1 particles (both infectious and non infectious) and signaling molecules on target and nontarget cell types. This interaction could potentially alter signaling events that trigger the release of proinflammatory cytokines and chemokines. Although HIV-1 interacts with several signaling molecules, in this study we focused on MAPKs, as these kinases are involved in multiple cellular functions, including activation, proliferation, differentiation, survival, and cytokine/chemokine production (Kyriakis and Avruch 2001). This is supported by a previous study that indicated that treatment of cells with ERK1/2 and JNK/SAPK inhibitors reduces MCP-1 and MIP-1 production (Kim and others 2006). We investigated whether HIV-1 infection could alter the phosphorylation of MAPKs such as ERK1/2, p38, or SAPK/JNK that could indirectly modulate CXCL5 production. We observed a similar correlation between CXCL5 and ERK1/2 signaling pathways, as blocking of ERK1/2 reduced CXCL5 level in HIV-1-infected macrophages compared with mock-infected control. Our data are consistent with the results shown in hepatocellular carcinoma patients that the overexpression of CXCL5 was associated with ERK1/2 phosphorylation (Zhou and others 2012). Although how the activation of ERK1/2 is related to CXCL5 production is not established; it is possible to predict that phosphorylation of ERK1/2 could phosphorylate or translocate transcription factors and/or IL-1β that could potentially bind and transactivate the CXCL5 promoter.
CXCL5 has been shown to have a role in CNS diseases, as increased levels of CXCL5 was observed in cerebrospinal fluid during the early hours of ischemic stroke (Zaremba and others 2006). Additionally, dysregulation of CXCL5 expression in response to HSV and JCV was observed in the CNS compartment (Aravalli and others 2005; Darbinyan and others 2013). Together, these studies established a role for CXCL5 in virus-induced CNS diseases. However, the effect of CXCL5 was not evaluated in the context of HIV-1 infection, although several studies suggested the involvement of CCL2 and CXCL10 (van Marle and others 2004; El-Hage and others 2006; Williams and others 2013) in HIV-1-induced neuropathogenesis. In this study, we demonstrate that HIV-1-induced CXCL5 does not have any direct effect on neuronal death/survival, as blocking of CXCL5 had no beneficial or adverse effect on neuronal survival. Although the consequence of increased CXCL5 in HIV-1-infected tissue compartment is not well established, secretion of CXCL5 from macrophages in the periphery and CNS could contribute to the infiltration of lymphocytes and neutrophils to the site of inflammation. It is known that ELR+ chemokines contribute to host defense through their involvement in leukocyte migration and activation. Our study and other previous studies have indicated that increased expression of CXCL5 has a direct chemoattracting effect on neutrophils in viral and nonviral diseases such as metastatic hepatocellular carcinoma as well as in hepatitis-infected mice with viral encephalomyelitis (Hosking and others 2009; Zhou and others 2012). To examine if CXCL5 has a role in infiltrating neutrophils and is not mediated only by the other chemoattractant chemokines, such as IL-8 (Harada and others 1994), neutralizing antibodies against CXCL5 were used. Our result confirmed that CXCL5 primarily contributes to the movement of neutrophils or CXCR2-positive cells. Contribution of CXCL5 in polymorphonuclear neutrophil infiltration has also been reported previously in pulmonary mycobacterial infections (Nouailles and others 2014). As HIV-1 infection is associated with immune activation and inflammation, it is possible to predict that the virus may have a role in infiltration of other cell types. The overproduction of proinflammatory cytokines such as IL-1β or TNF-α during HIV-1 infection could result in transcriptional activation of CXCL5 in resident macrophages/microglia that in turn recruits the neutrophils toward the inflamed tissues. Hyperactivation of neutrophils in HIV-1-infected patients is associated with deregulation of the apoptosis/necrosis equilibrium (Campillo-Gimenez and others 2014). Thus, understanding the inflammatory process of HIV-1-infected target cells is an important aspect of HIV-1 biology, especially in the tissue compartments where accumulation of cellular factors remains high. These factors, depending on the amount and its interaction with the surrounding cell types, may result in deleterious effect and tissue damage as observed in brain tissue compartments of HIV-1-infected individuals. Defining the inflammatory networks at the lymphoid and CNS compartment and its downstream effects may provide new avenues for immune therapeutic options to relive the inflammatory burden and its biological consequences.
Footnotes
Acknowledgments
This work was supported by R01 MH087247 to V.A. from the NIMH, NIH, and D.G. was, in part, supported by the AIDS International Training grant D43TW001038. The authors thank Prof. Lluis Fajas Coll, Switzerland for the CXCL5 promoter.
Author Disclosure Statement
The authors declare that they have no competing financial interests.