
Abstract
Type 1 interferons (IFNs) have been shown to be efficacious against hepatitis C virus (HCV), hepatitis B virus (HBV), and some cancers with a significant drawback of short drug exposure. We have significantly improved the pharmacokinetic (PK) of consensus interferon (CIFN) by glycoengineering. We generated AL-624 by introducing 4 glycosylation sites. AL-624 was expressed, purified, and fractionated to yield 2-Gly, 3-Gly, and 4-Gly. In a rat PK study, AL-624 4-Gly exhibited a 6-fold increase of area under curve (AUC) and more than an 11-fold increase in time to half life (T1/2) over CIFN, suggesting the potential for weekly dosing (QW). In Yellow fever virus hamster model, QW of 4-Gly achieved similar efficacy to daily dosing (QD) CIFN and QW Peg-IFN-α-2a in overall survival rate and reduction of alanine aminotransferase (ALT) level. Further refinement resulted in development of AL-683 by addition of external glycosylation sites and its mouse homologue. AL-683 maintains undiminished biological potency in HCV replicon. In mouse PK/pharmacodynamic (PD) studies, AL-683 homologue has a ∼37-fold improvement in T1/2 and a ∼33-fold improvement in AUC when compared with the unglycosylated mouse IFN-α-1. Significantly improved PD responses were also observed. The significant improvement of AL-683 PK over AL-624 suggests a bimonthly dosing regimen for AL-683. The possibility for once-a-month dosing could be realized by further optimization of manufacturing conditions.
Introduction
I
Pegylation, a chemical transformation that attaches polyethylene glycol (PEG) moieties, has been successfully applied to improve PK properties of many proteins such as IFN-α-2a and IFN-α-2b (Kozlowski and others 2001). The increased molecular weight (MW) and hydrodynamic volume of these pegylated interferons reduces their renal clearance, increases their serum half life, and leads to a significant elongation of the biological effect. As a consequence, the dosing frequency of the pegylated IFN-α-2a (Pegasys®; Roche, Basel, Switzerland) and pegylated IFN-α-2b (PegIntron®; Merck, Whitehouse Station, NJ) is reduced from daily dosing (QD) to weekly dosing (QW). However, pegylation also has its drawbacks, including a major loss of biological activity in products such as pegylated IFN-α-2a (Bailon and others 2001) and a cost of goods increase due to complex postproduction modification and purification procedures. To preserve maximum biological activity, pegylation typically adds mono 10–40 kD PEG chains to proteins, which, in most cases, will not extend dosing beyond once per week.
Protein glycosylation engineering provides a viable alternative in improving the PK profile of protein therapeutics. Glycosylation is one of the most common protein modifications in eukaryotes. There are 2 types of glycosylation: N-linked and O-linked. The core of the N-linked glycosylation motif is N-X-T/S, where N (Asparagine) is the anchoring amino acid for the carbohydrates; X can potentially be many different amino acids; and the third position is either T (Threonine) or S (Serine). The core sequence of O-linked glycosylatyion is less defined; however, the anchoring residues are most commonly S or T. Glycosylated molecules may acquire significantly improved plasma half life by increasing MW and hydrodynamic radius against renal filtration, reduced proteolysis by carbohydrate chains blocking protease access to sensitive regions, and lowered immunogenicity by masking the epitopes. Furthermore, glycosylation eliminates the need for chemical processing postprotein production. Unlike pegylation, carbohydrates are added to precise amino acids during fermentation in a glycoengineered protein therapeutic. Glycoengineering has been successfully applied in the development of Aranesp® (manufactured by Amgen, Thousand Oaks, CA), a recombinant erythropoietin (EPO) derivative indicated for use in the treatment of anemia (Elliot and others 2003). Aranesp can achieve once-a-week dosing compared with thrice-a-week dosing for the prototype EPO.
Currently, no glycoengineered drug has achieved dosing frequency less than once per week. Pegylated IFN-α-2a and IFN-α-2b are also dosed at once per week. In this article, we report a glycosylation platform technology and its application to IFN alfacon-1 (Infergen®; Kadmon Corp., New York, NY) a.k.a. consensus interferon (CIFN), which leads to greatly improved PK properties, allowing for bimonthly or monthly dosing while maintaining the highly potent biological activity of the parent CIFN.
There are a large number of subtypes for human IFN-α, including IFN-α-2a and IFN-α-2b. CIFN is a synthetic interferon derived from the consensus amino-acid sequences from 8 natural subtypes (Ozes and others 1992). It has been approved in treating CHC infections. However, due to a poor PK profile, it requires daily dosing. Compared with 2 leading drugs in hepatitis C virus (HCV) treatments, pegylated IFN-α-2a and IFN-α-2b, CIFN exhibits increased anti-HCV potency and a distinct interferon stimulated gene pattern (Erickson and others 2008). This could be the reason underlying several clinical findings that CIFN can be a therapeutic alternative to pegylated IFN-α-2a and IFN-α-2b for their nonresponders or relapsers (Heathcote and others 1998). However, its frequent dosing severely hampers the potential of this drug. For CIFN to become a true alternative or best in class drug, its PK parameter has to be improved to allow dosing once per week or even longer. Our eventual goal is have a once-per-month dosing drug in treating chronic hepatitis B virus (HBV), HCV infections, or other emerging viral diseases, which will distinguish themselves from other pegylated IFN drugs. Once a month, injections could be administered in a doctor's office during visits and, therefore, increase the patient compliance and treatment outcome, along with the inherent advantage of further reduced dosing. In the era of direct-acting antiviral agents (DAAs) in HCV treatments, the use of all oral IFN-free regiments have now become the Standard of Care and IFN is no longer the main treatment option for many patients. However, several studies have demonstrated the preventive effects of IFN in CHC patients from developing hepatocellular carcinoma, and low-dose IFN maintenance therapy might still be beneficial in certain populations (Enomoto and others 2014). For these populations and for patients who cannot afford the cost of DAA, once-a-month dosing IFN could potentially offer benefits. IFNs have also been approved for treating CHB, and this is the area where once-a-month IFN may be the most beneficial to patients. DAAs are also available in treating chronic HBV infection; however, the treatment is usually lifelong and long-term toxicity and emergence of viral resistance mutations can be a problem. Despite its side-effects, IFN is still considered one of the main treatment options in CHB and pegylated IFN can achieve an overall sustained response rate of ∼30% (Asselah and others 2007).
We began the project by designing the glycosylation sites in CIFN utilizing the homologous amino-acid sequence alignments with other human Type I IFN species with naturally occurring glycosylation sites. We demonstrated a significantly improved PK with 4 glycosylation site additions (4-Gly) and showed efficacy in Yellow fever virus (YFV) hamster model (Julander and others 2007) with once-a-week dosing, compared with daily dosing for the parent molecule. However, as with the pegylation of IFN-α-2a, the 4-Gly CIFN suffers significant loss of biological activity. By reducing the number of internal glycosylation sites from 4 to 2, and by adding 14 external glycosylation sites to the N-terminal end of CIFN, we were able to generate a hyperglycosylated CIFN with MW 70–100 kD targeting once a month dosing and no loss of biological activity. Due to the lack of available rodent model to test the efficacy of once-a-month dosing IFN, we chose to run a PK/pharmacodynamic (PD) study in the mouse to demonstrate the value of our core glycosylation technology. Human IFN cannot induce a PD response in mouse due to species difference; thus, a hyperglycosylated mouse homologue of human Type I interferon was generated with 14 glycosylation sites at N-terminal of the protein. The PK and PD properties of this homologue were analyzed in a mouse study. Further significant improvement in PK profile over the previous 4-Gly species demonstrated that this technology is capable of overcoming some of the limitations of Pegylation to yield highly active and long-lasting glycosylated CIFN targeting bimonthly or monthly dosing. However, there is still limitation in extrapolating murine IFN result to that of human IFN. The efficacy of the human IFN targeting once-a-month dosing can be tested more appropriately in certain primate models (Bukh 2004), even though there is a tremendous amount of financial, regulatory, and ethical challenges in using such models.
Materials and Methods
Growth medium and reagents
FreeStyle 293-F cells (Invitrogen, Carlsbad, CA) were used for the expression of glycosylated proteins. FreeStyle 293-F cells were cultured in ready-to-use Gibco FreeStyle 293 Expression Medium with the addition of 1% penicillin-streptomycin. The cells were grown in suspension, rotating in an orbital shaker at 135 rpm and incubated at 37°C with 8% CO2.
Molecular cloning and mutagenesis
To express CIFN in mammalian cells as secreted protein, its codons were optimized for mammalian expression and the signal peptide of human IFN-α-14 was fused to the N-terminal of the protein. CIFN and hyperglycosylated CIFN with external glycosylation sites were de novo synthesized (IDT, Coralville, IA) and cloned into pcDNA3 (Invitrogen). The CIFN mutants with internal glycosylation sites were generated with the QuikChange™ Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA).
Transient transfection
Approximately 24 h before transfection, FreeStyle™ 293-F cells were passed at 6–7×105 cells/mL. On the day of transfection, 30 mL of cells (1×106 cells/mL) were added into each 125 mL shake flask. In 2 separate tubes, 37.5 μg of plasmid DNA and 37.5 μL of FreeStyle MAX Reagent were diluted into OptiPro™ SFM to a volume of 0.6 mL each. Then, the diluted FreeStyle MAX Reagent was added to the diluted DNA solution to obtain a total volume of 1.2 mL. The DNA-lipid mixture was incubated for 10 min at room temperature to allow complexes to form. 1.2 mL of DNA-lipid mixture was added drip-wise into the 125 mL flask containing cells while slowly swirling the flask. The transfected cell cultures were incubated at 37°C, 8% CO2 on an orbital shaker platform rotating at 135 rpm for 3–5 days. For larger-scale production of glycosylated proteins, the cells, plasmid DNA, and FreeStyle Max reagent were scaled up proportionally.
Protein purification of AL-624: CIFN with 4 internal glycosylation sites
AL-624 was collected from transfected FreeStyle 293-F cells. It was first concentrated by methods previously described (Zaworski and Gill 1988). Zinc chloride was added to the cell supernatant to a concentration of 5 mM. NaOH was added to adjust the pH to 7.4. At this point, a precipitate was formed, collected by centrifugation, and discarded. Zinc chloride was added again, now to 30 mM. NaOH was added again to achieve pH 7.4. A second precipitate was formed and collected by centrifugation. This precipitate was resuspended with 0.5 M EDTA, 1 M Tris pH 7.5 and concentrated using a Jumbo-Sep concentration unit (Pall Corp. Port Washington, NY) with a 10,000 MW cutoff membrane. Protein was further purified by size exclusion chromatography using a Superdex 200 26×60 column in TBS at pH 7.4. Column fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Fractions were concentrated using Vivaspin centrifugal devices with a 10,000 MW cutoff membrane. Protein concentration was determined by Bradford assay.
Enzymatic deglycosylation
When needed, the added sugar chains from our glycosylated CIFN were removed using a PNGase F kit (New England Biolabs, Ipswich, MA), following the manufacturer's instructions. In brief, 1–20 μg of glycoprotein was added to the Glycoprotein Denaturing Buffer and denatured by heating the reaction at 100°C for 10 min. Then, G7 Reaction Buffer, NP-40, and PNGase F were added and incubated at 37°C for 1 h.
CIFN sandwich ELISA
To determine the quantity of CIFN or its glycosylated analogs in conditioned media, a sandwich ELISA was used. The glycosylated CIFN analogs were deglycosylated first before being subjected to ELISA testing. The CIFN manufactured for clinical use (Infergen; 30 μg/mL) was used as the standard in each ELISA. In brief, the rabbit polyclonal anti-CIFN antibody was used as capturing antibody and the same antibody that had been biotinylated was used as detection antibody. After complex formation and multiple washing steps, the streptavidin-conjugated horseradish-peroxidase (GE Life Sciences, Piscataway, NJ) was added and the standard ELISA procedure was followed.
Hepatitis C virus replicon assay
An HCV subgenomic replicon system, derived from a hepatoma cell line Huh7, expressing a Firefly luciferase-encoding HCV genotype 1b replicon was used (Bartenschlager and others 2003). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 4,500 mg/L
On the day before dosing, 5,000 HCV replicon cells per well were plated in a 96-well plate to grow overnight. On the next day, each protein was serially diluted (1:3) to 9 distinct concentrations, added to the replicon cells in a 96-well plate, and further incubated for 3 days. Finally, the Bright-Glo Luciferase Assay System (Promega, Madison, WI) was used to measure anti-HCV replicon activity.
Rat PK study
In vivo PK testing was performed using 12 Sprague–Dawley rats (each 275–300 g in size), 3 rats per group for 4 test articles. They were dual cannulated at the right jugular vein and left carotid artery. Each rat received a single dose of 2×105 international unit (IU) of interferon subcutaneously. The test articles were divided up between different interferons: CIFN, 2-Gly, 3-Gly, and 4-Gly. The dose was delivered at 300 μL in a vehicle of TBS. One hundred to 125 μL of plasma was collected at each of the 14 time points: 5, 15, and 30 min; and 1, 2, 3, 4, 6, 8, 10, 24, 48, 72, and 96 h postdose. The biological activity of human interferon was measured with the method shown in the next section. The PK parameters were calculated using WinNolin (Pharsight, Sunnyvale, CA).
Measure CIFN in animal plasma
Measurement of interferon biological activity in animal plasma was performed using iLite alphabeta Human Type I Interferon Activity Detection Kit produced by PBL (Piscataway, NJ) following the manufacturer's instructions. In brief, the plasma samples containing CIFN and its analogs, as well as a serially diluted Type I interferon standard provided in the kit, were added to the interferon-sensitive cells. These cells are capable of generating luciferase when the interferon signal pathway is turned on. After incubation and addition of lysis reagent/substrate, the resultant bioluminescence intensity is proportional to the amount of Type I interferon activity (IU/mL) in the sample.
Yellow fever study
We conducted a study evaluating the effect of different interferon therapies against a hamster model of the Yellow fever disease. Jimenez, a hamster-adapted YFV strain, was obtained as a generous gift from Robert B. Tesh (University of Texas Medical Branch, Galveston, TX). The virus was inoculated into 5 adult female hamsters. The livers of the infected hamsters were removed 3 days postviral injection and homogenized in a 2×volume of sterile phosphate-buffered saline (PBS). This liver homogenate had a titer of 106.0 50% cell cultures infectious doses/mL (CCID50).
Female Syrian golden hamsters with an average weight of 110 g were randomly assigned to groups of 10–15 hamsters in each. A 10−4 dilution (102.0 CCID50/mL) of the virus was prepared in minimal essential media. Hamsters were injected intraperitoneally (i.p.) with 0.1 mL of the diluted virus (10 CCID50/animal). All compounds were administered i.p. at a dose of 1.25×106 IU/kg/dose, given once a week (QW, AL-624 4-Gly and Pegasys, −4 h and 7 days), twice weekly (BIW, AL-624 3-Gly, −4 h, 3.5, 7, and 10.5 days), or every day (QD, IFN alfacon-1 and placebo) beginning at 4 h and ending on 14 days postinfection (dpi). Mortality was observed daily for 21 days, and serum from each live hamster was collected on 6 dpi for alanine aminotransferase (ALT) level measurement.
Statistical analysis
Survival data were analyzed using the Wilcoxon log-rank survival analysis, and all other statistical analyses were done using 1-way ANOVA using a Bonferroni group comparison (Prism 5; GraphPad Software, Inc., La Jolla, CA).
Protein purification of AL-683 mouse homologue: His-tagged mouse IFN-α-1 with 14 N-terminal glycosylation sites
Ni-NTA Spin Kit columns (Qiagen, Hilden, Germany) were used for His-tag protein purification. The resins were taken from a single column, added to the 250 mL collected transfection supernatant, and incubated overnight with shaking at 4°C. Scale up can be done proportionally. After incubation, the resins were repacked into the column and washed 5 times each with a phosphate buffer solution containing 20 mM imidazole. They were then eluted with 0.5 mL of a phosphate buffer solution containing 500 mM imidazole. All passes through the columns were performed by gravity. The eluted protein was dialyzed in 1×PBS to eliminate the imidazole and then concentrated using Vivaspin centrifugal devices with a 10,000 MW cutoff membrane. Protein concentration was determined by Bradford assay.
Encephalomyocarditis virus cytopathic effect assay in mouse L929 cells
To measure the specific activity of AL-683 mouse homologue, an encephalomyocarditis virus (EMCV) cytopathic effect (CPE) assay was used with purified mouse IFN-α-1 (PBL) as standard. Each batch of mouse IFN-α-1 was supplied with a measured specific activity from the vendor. The L929 cells (ATCC, Manassas, VA) were cultured in DMEM containing 4,500 mg/L
AL-683 mouse homologue PK/PD study
Two groups of male C57BL/6 mice were dosed with the test article (AL-683 mouse homologue) and a control (mouse IFN-α-1), respectively. For the test article group, 30 mice were dosed subcutaneously with 1,000 IU/g AL-683 mouse homologue. Three mice were sacrificed to collect plasma and livers at each of the following time points postdosing: 0, 0.5, 1, 3, 5, 12, 24, 48, 96, and 168 h. For the control group, 18 mice were dosed subcutaneously with 1,000 IU/g mouse IFN-α-1. Three mice were sacrificed to collect plasma and livers at each of the following time points postdosing: 0, 12, 24, 48, 96, and 168 h. The mouse interferon activity in plasma was measured with VeriKine™ Mouse Interferon Alpha ELISA Kit (PBL) following the manufacturer's instructions. Conversions of concentrations of test article and control in mouse plasma from ng/mL to IU/mL were based on the specific activity of each protein, respectively.
Mouse β2-microglobulin level in plasma was measured with an ELISA kit (USCN Life Science, Wuhan, China) following the manufacturer's instructions. Total RNA was isolated from the preserved mouse liver using the RNeasy Mini Kit (Qiagen). Mouse 2′-5′-oligoadenylate synthetase 1 (OAS1) mRNA level was quantified with real-time PCR using the following primer/probe set: Forward primer, reverse primer, and probe have the following sequences, respectively: GCT GAC CTG GTG GTG TTC CTT; CCC GTC GGT TTA ACT GAT CCT and 6FAM-ACA ATC TCA CCA GCT TT-MGBNFQ. The levels of liver OAS1 were normalized against the levels of endogenous GAPDH, measured with real-time PCR using Mouse GAPDH Endogenous Control Kit (Invitrogen).
Results
Design CIFN internal glycosylation sites
The amino-acid sequence of CIFN was aligned with other naturally glycosylated human Type I interferons, and the CIFN glycosylation sites were designated at or near the corresponding amino-acid positions (Fig. 1A). P4N, D71N, and D72N are at or near the corresponding positions of IFN-α-14 glycosylation sites (Zhao and others 1987). D78N is at the corresponding position of IFN-β and IFN-ω1 glycosylation sites (Conradt and others 1987; Runkel and others 1998). E107T and V114N are at or near the corresponding position of IFN-α-2b glycosylation site (Nyman and others 1998). All of the designed sites are N-linked glycosylation sites, expect for E107T, which is an O-linked glycosylation site.
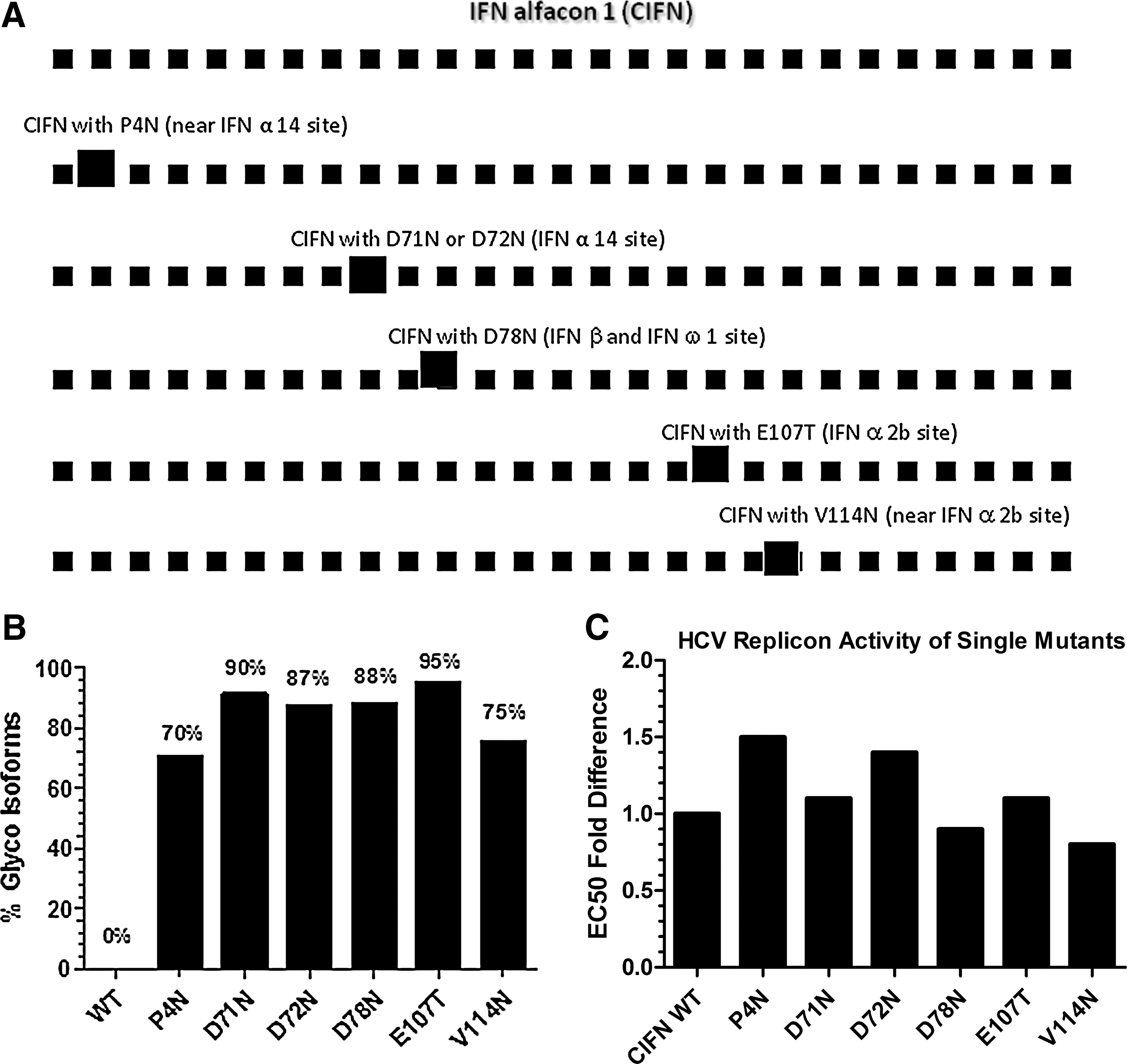
The N-linked glycosylation sites in consensus interferon (CIFN) molecules were designed at or near the naturally occurring glycosylation sites in homologous Type I human interferons. E107T is an O-linked glycosylation site, and the rest are N-linked glycosylation sites
Glycosylation efficiency and effect on biological activities
Mammalian codon optimized CIFN was de novo synthesized and cloned into mammalian expression vector. The single glycosylation mutations outlined were generated individually to assess the glycosylation efficiency at different locations and the effect of the glycosylation on biological activity. All designed sites yielded a high degree of glycosylation (>70% efficiency), especially those sites (D71N, D72N, D78N, and E107T) that are closest in the alignment with the naturally occurring glycosylation sites (Fig. 1B).
In the HCV replicon assay, all of the single glycosylation mutants behaved similar to the wild-type nonglycosylated CIFN with a difference in EC50 being smaller than the 2-fold change that falls within the assay variation (Fig. 1C).
Generation of once-a-week dosing CIFN AL-624
AL-624 with glycosylation sites at 4, 71, 78, and 114 was generated by site-directed mutagenesis. A Homology Model of AL-624 was generated and was overlaid with an NMR-derived model of human IFN-α bound to extracellular domain of IFNAR2 receptor (R2-EC) (Quadt-Akabayov and others 2006), allowing us to assess the positions of the mutations in 3D. The model shows that the engineered glycosylation sites at 4, 71, 78, and 114 are located in flexible loops or helical junctions, away from helix bundle packing and receptor-binding interfaces (Fig. 2A). Sugar chains were built explicitly at the glycosylation sites, and low-energy conformations were explored using molecular mechanics energy minimization, as implemented in MacroModel™ (Schrödinger, LLC, New York, NY). These calculations showed that sugar chains introduced at sites 4, 71, 78, and 114 adopt conformations that keep them away from unfavorable overlaps with CIFN AL-624 protein packing interfaces. Molecular Dynamics simulations (DESMOND™; D. E. Shaw Research, LLC, New York, NY) were carried out on a number of glycosylated CIFN variants to assess the effect of glycosylation on overall protein fold and hydrodynamic radius.

Three-dimensional model of AL-624 and extracellular domain of IFNAR2 receptor (R2-EC)
The plasmid with AL-624 DNA under the control of CMV promoter was transiently transfected into FreeStyle293 cells, and the expressed and secreted AL-624 protein was purified from the media and fractionated by their sizes. The fractions were analyzed by SDS-PAGE followed by Commassie Blue staining (Fig. 2B). Among these fractions, 3 were chosen for further analysis and their median sizes are around 40, 36, and 30 kD, respectively. They were named 4-Gly, 3-Gly, and 2-Gly fractions, because their sizes reflect CIFN variants with 4, 3, and 2 fully glycosylated sites. It was demonstrated that the increase in MW of these fractions from unglycosylated CIFN control was the consequence of protein glycosylation. When the sugar chains were removed by enzymatic deglycosylation, the sizes of the CIFN variants were decreased to the same as CIFN control (Fig. 2C). When biological activities of these fractions were analyzed (Fig. 2D), there was an incremental decrease of biological activity along with a higher degree of glycosylation: 4-Gly EC50>3-Gly EC50>2-Gly EC50. The potency of 2-Gly is similar to that of wild-type CIFN, while 4-Gly is similar to PEG-IFN-α-2a (Pegasys).
PK results of AL-624 in rats
The PK parameters of the fractions of AL-624 along with CIFN control in rats are shown in Table 1. The human interferon activity was measured for each collection, and the results were plotted (Fig. 3). All of the fractions of AL-624 have a longer time to half life (T1/2) and a higher area under curve (AUC) when compared with the unglycosylated CIFN. When the fraction has a higher degree of glycosylation, it performs better in its PK profile. The AUC to AL-624 4-Gly was 6-fold higher than that of CIFN. The T1/2 of AL-624 4-Gly increases more than 11-fold compared with CIFN.
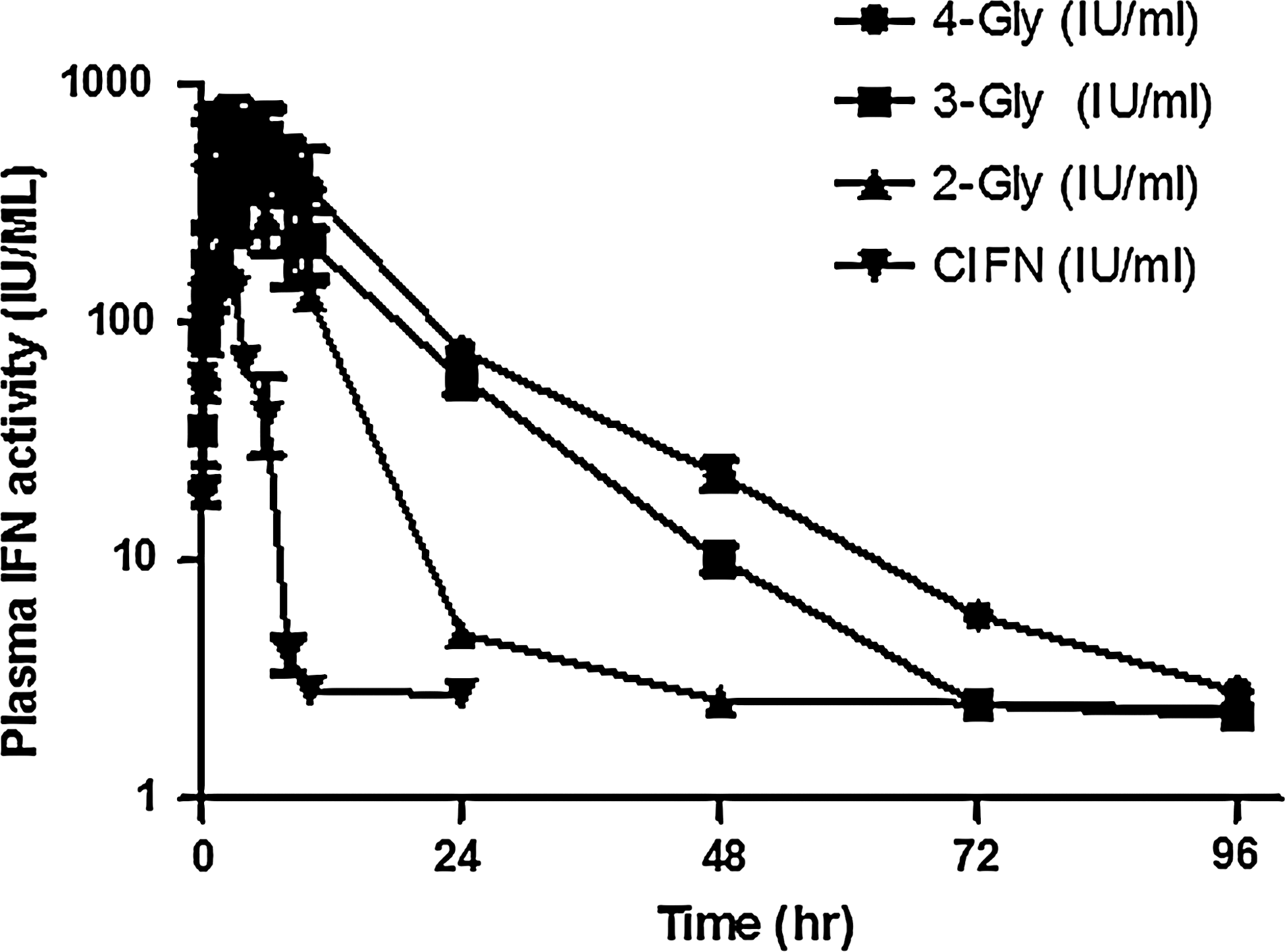
Pharmacokinetic (PK) study of AL-624 in rats with single subcutaneous dose.
AUC, area under curve; CIFN, consensus interferon; T1/2, time to half-life.
AL-624 efficacy study in YFV hamster model
The primary end point was to compare the overall survival rate between different groups (Fig. 4A). When dosed once a week in YFV-infected hamsters, AL-624 4-Gly achieved 90% survival rate at the end of the study. It was identical to the survival rate for the once-a-week pegylated IFN-α-2a (Pegasys) group. The once-a-day CIFN group achieved the highest survival rate: 100%, but there was no statistical difference between this and either AL-624 4-Gly or Pegasys group. Twice-a-week dosing of AL-624 3-Gly had a survival rate of 80%. For the placebo group, 1× PBS was dosed daily and the survival rate for placebo group was 20%. All 4 treatment groups achieved statistically significant survival benefit (P<0.01) when compared with the placebo group.

AL-624 efficacy study in Yellow fever virus (YFV) hamster model. The survival rate is the primary end point. The effects of AL-624 4-Gly, 3-Gly, CIFN, and pegylated IFN-α-2a (Pegasys) on survival in hamsters infected with YFV (***P<0.001, **P<0.01, as compared with placebo) are shown
When the serum ALT levels of the treatment groups were compared with the placebo group (Fig. 4B), AL-624 4-Gly weekly dosing (P<0.01), Pegasys weekly dosing (P<0.05), and CIFN daily dosing (P<0.01) achieved statistically significant reductions in ALT levels. However, the reduction of ALT in AL-624 3-Gly group was not statistically significant.
Design of hyperglycosylated CIFN targeting once-a-month dosing
As demonstrated by the rat PK study and YFV hamster study, an addition of 4 glycosylation sites to the native sequence of CIFN will yield a molecule sufficient for once-a-week dosing. To target once-a-month dosing, significantly more glycosylation sites are needed in each molecule. However, cell biology data showed that the higher degree of glycosylation in CIFN will lead to more loss of biological activity. Along with addition of 4 substantially glycosylated sugar chains (AL-624 4-Gly), CIFN is ∼100-fold less potent than the wild-type CIFN. We faced a seemingly insurmountable challenge in our efforts to drastically improve the PK profile while maintaining majority of biological activities in wild-type unglycosylated CIFN. A creative method of adding large numbers of glycosylation sites to the N-terminal of CIFN was formulated. After many exploratory studies (data not shown), AL-683 (Fig. 5A) was created as our lead candidate in targeting once-a-month dosing. AL-683 has incorporated 14 N-terminal glycosylation sites by adding an artificial sequence between the signal peptide and CIFN regions. The artificial sequence consists of 14 N-linked glycosylation sites as well as 3 Glycine and 1 Valine residues (GGGV) as a linker between 2 sites. Glycine residue was chosen for its small side chain and lower steric hindrance for the sugar chains. Besides the external 14 glycosylation sites, ALS-683 retains on internal glycosylation site D78N.
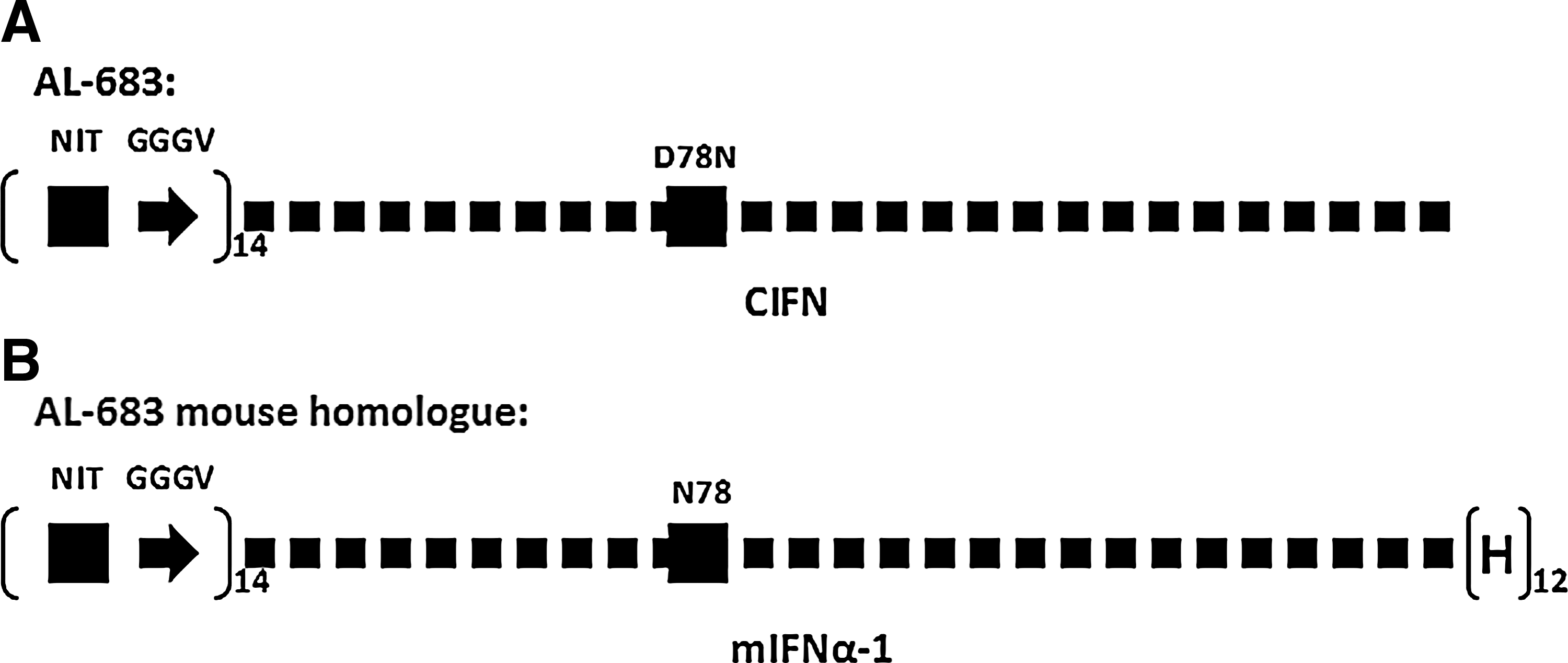
AL-683 was designed to add 1 internal and 14 external glycosylation sites to CIFN. Site-directed mutagenesis was carried out to generate a glycosylation site internally at amino-acid position 78. The external engineered glycosylation site (NIT) and the linker region (GGGV) have been repeated 14 times at the N-terminal of CIFN
AL-683 has large MW and undiminished biological activity
AL-683 was transiently over-expressed in FreeStyle 293 cells, and the secreted form in the conditioned media was analyzed for its size by gel electrophoresis and Western blot (Fig. 6A). The MW range of the AL-683 is 70–100 kD and the medium size is similar to that of Albuferon (85.7 kD), which was tested for once-a-month dosing in human clinical trials (Osborn and others 2002). The AL-683 protein concentration in the conditioned media was determined by sandwich ELISA after enzymatic deglycosylation. The HCV replicon assay was used to measure the antiviral activity of hyperglycosylated AL-683 as well as the unglycosylated CIFN (Fig. 6B). The EC50 of AL-683 is 1.2-fold that of CIFN within the 3-fold range that is typical of assay-to-assay variation. Thus, AL-683 has a ∼4-fold increase in MW when compared with CIFN yet it maintains the same level of biological activity as its parent.
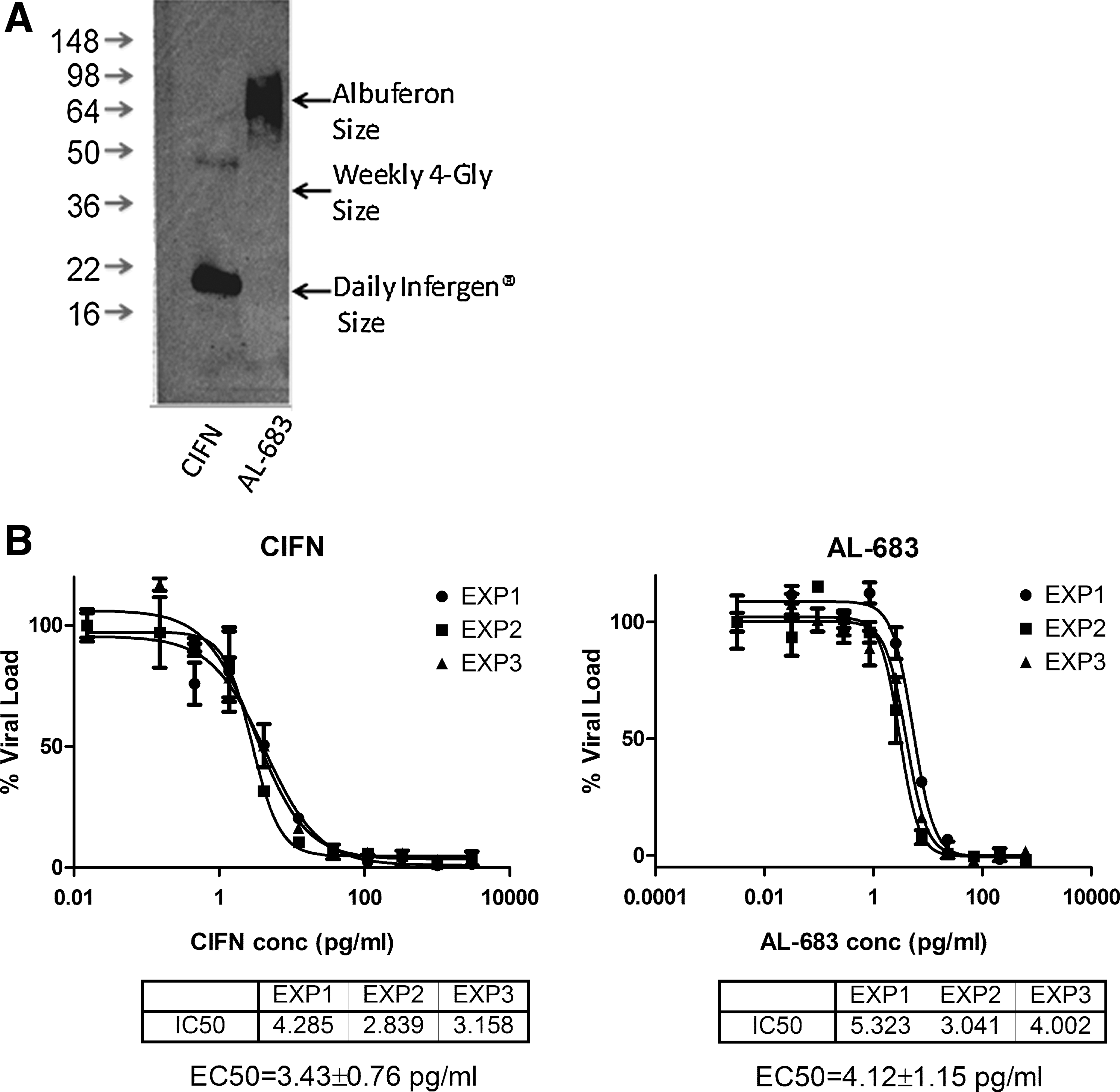
AL-683 was transiently expressed in FreeStyle 293-F cells, and size was visualized by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot with anti-CIFN polyclonal antibodies
AL-683 mouse homologue generation and PK/PD study
It is our interest to understand the PD effect of the longer-lasting CIFN in patients, and the first proof-of-concept study was conducted in the mouse. Due to species differences in IFN binding to its respective receptor, we constructed a mouse homologue of AL-683 using murine IFN-α-1 (Fig. 5B). Murine IFN-α-1 was chosen among murine Type I IFNs, because it was well studied and showed PD effects in the mouse (Sarasin-Filipowicz and others 2009). The signal peptide is the native signal peptide of mouse IFN-α-1. The sequence for external extension of glycosylation sites is identical to that of AL-683. No internal glycosylation site was generated, because there is 1 natural glycosylation site in native mouse IFN-α-1. Thus, the overall glycosylation site number is identical between AL-683 and its mouse homologue. A total of 12 Histidine residues were added to the C-terminal of the protein to facilitate the protein purification.
AL-683 mouse homologue was transiently transfected into FreeStyle 293-F cells and the expressed, His-tagged, secreted fusion protein was purified with Ni-resins. The eluted protein was evaluated with SDS-PAGE and then Coomassie Blue staining (Fig. 7). The protein was most abundant in elution fraction number 2, and the size of the protein was 90–100 kD. The protein was then dialyzed and concentrated. The protein concentration (mg/mL) was measured with the Bradford method, and protein specific activity (IU/mg) was determined with the EMCV CPE assay.
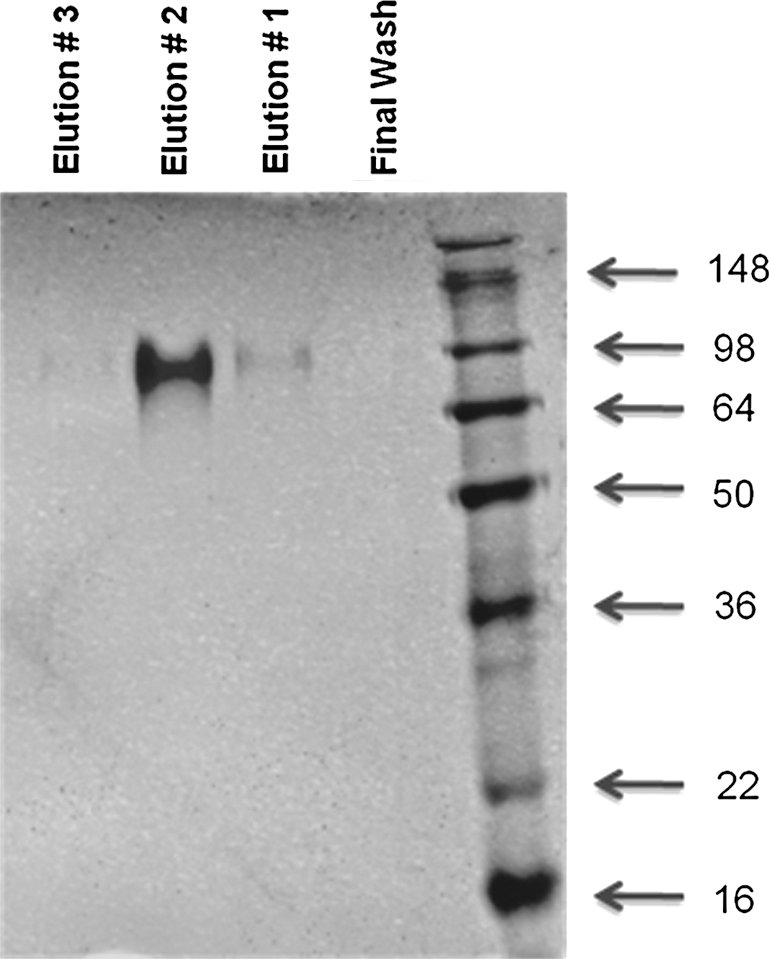
AL-683 mouse homologue was transiently expressed in FreeStyle 293-F cells and purified through Ni-resins. The elution fractions were analyzed through SDS-PAGE and Coomassie Blue staining.
In the mouse PK/PD study, the dose for both AL-683 mouse homologue and unglycosylated mouse IFN-α-1 control was 1,000 IU/g. The plasma mouse IFN-α-1 activity per milliliter with the progression of time is shown in Fig. 8. The calculated PK parameters are summarized in Table 2. Overall, there was a significant improvement in the PK profile of mouse IFN-α-1 when 14 external glycosylation sites were added. AL-683 mouse homologue showed a ∼37-fold improvement in T1/2 and ∼33-fold improvement in AUC when compared with unglycosylated mouse IFN-α-1.
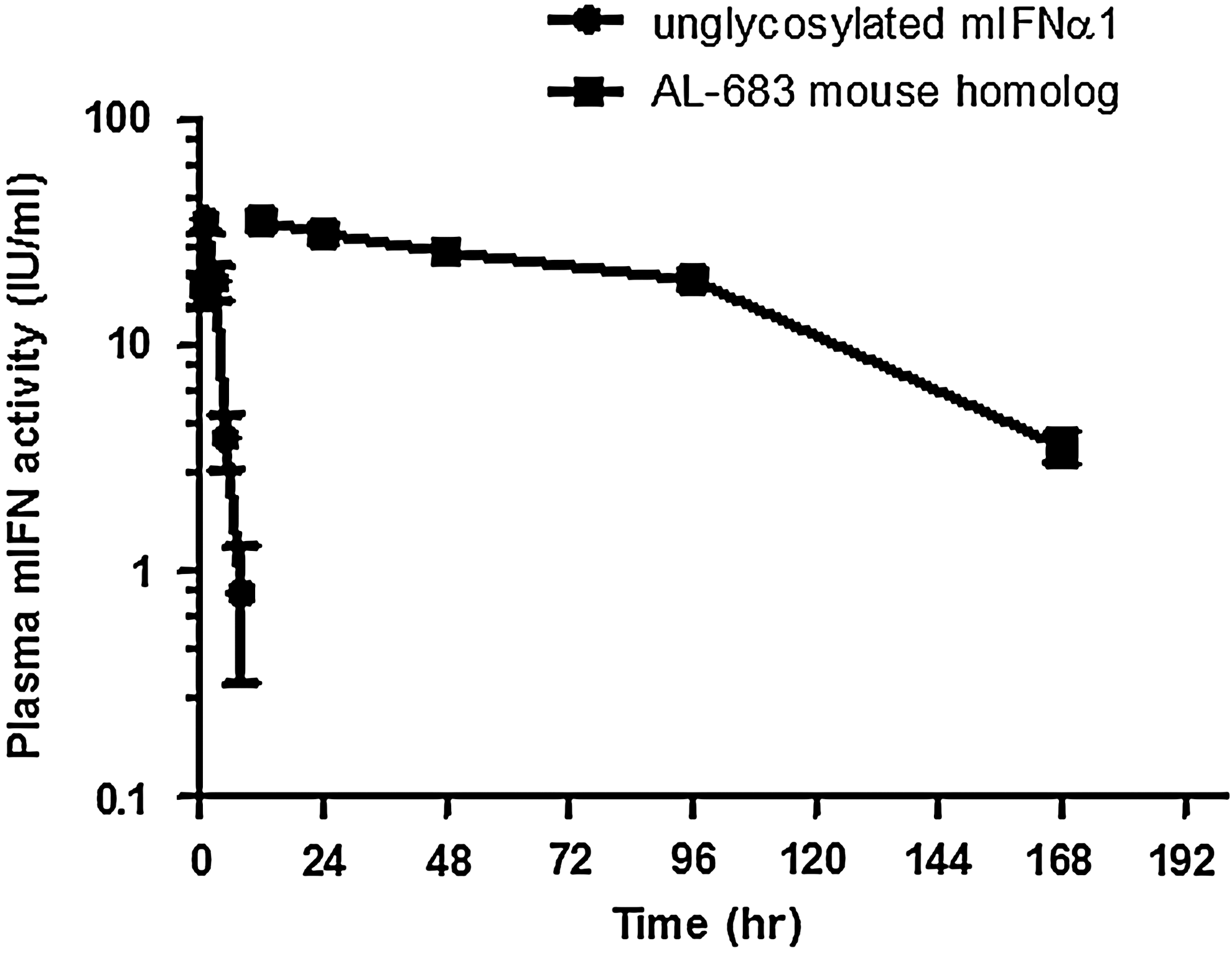
AL-683 mouse PK study: plasma mouse interferon activity versus time.
In the mouse PD marker time course study, β2 microglobulin level in the plasma was measured over a 1 week period (Fig. 9A). For the control group (mouse IFN-α-1), the protein peaked at 24 h postdosing and dropped down to baseline level at 48 h postdosing. In AL-683 mouse homologue group, the protein peaked at 24–48 h and took about 1 week to drop down to baseline level. Its AUC is roughly 2.6-fold more than that of the control group. We had difficulty in detecting the OAS1 protein with commercially available ELISA kits due to sensitivity issues and decided rather to measure the induction of messenger RNA level in the liver (Fig. 9B). The OAS1 mRNA level was normalized against mouse GAPDH mRNA level. In the mouse IFN-α-1 control group, the induction of OAS1 mRNA peaked at 12 h postdosing and returned to baseline level at 24 h postdosing. For the AL-683 mouse homologue group, the OSA1 mRNA peaked between 12 and 24 h postdosing and dropped down to near baseline during the 1 week period. The AUC difference between 2 groups was 3-fold.

AL-683 mouse pharmacodynamic study: plasma β-2 microglobulin protein-fold increase over baseline versus time
Discussion
Unique method of designing glycosylation sites within CIFN
There are more than a dozen known human Type I interferon subtypes (Pestka 2007), and some of them have naturally occurring glycosylation sites. These subtypes have a high degree of amino-acid sequence homology with each other and CIFN. After a sequence alignment of CIFN with the naturally glycosylated Type I human IFNs, glycosylation sites can be designed in CIFN at or near the homologous sites. These sites were selected through evolution, and their glycosylations most likely will not dramatically affect the protein activities. It has also been reported that some of these naturally occurring glycosylations play a role in maintaining the biological activities of some IFNs (Utsumi and others 1995; Runkel and others 1998). Designing the glycosylation sites at or near homologous sites will likely yield variants with high glycosylation efficiency and little biological activity reductions; therefore, this will significantly reduce the work load compared with scanning through the whole protein to identify glycosylation sites that might maintain the biological activities. Adopting the strategy of designing glycosylation sites around the native sites in the homologous species, we indeed achieved a very high success rate in identifying glycosylation sites in CIFN that are both efficiently glycosylated and highly active.
Design and generate once a week dosing CIFN
Peginterferon alfa-2b has been approved for once-a-week dosing with an average MW of 31 kD, and Peginterferon alfa-2a has also been approved for once-a-week dosing with an MW of 60 kD (Kozlowski and others 2001). To achieve once-a-week dosing for CIFN, 40 kD was our initial targeted MW. It was estimated that each N-linked glycosylation added roughly 5 kD to the parent EPO molecule (Elliot and others 2003). To reach 40 kD MW, at least 4 glycosylation sites were necessary for CIFN, which has an original MW of 19.4 kD. It has also been reported that 4 glycosylation sites in human IFN-α-2b showed a likely PK profile of once-a-week dosing (Ceaglio and others 2008).
A few CIFN variants with various combinations of 4 sites selected from the 6 single sites described were generated and examined for efficiency of glycosylation; AL-624 with glycosylation sites at 4, 71, 78, and 114 was found to be slightly better than the others and was chosen for further development (data not shown). A 3D model also showed favorable locations of these sites, away from blocking the interactions between IFN and its receptor. Even so, we still saw a gradual decrease of biological activity with a higher degree of glycosylation. The highest glycosylated 4-Gly fraction of AL-624 has more than a 100-fold loss of biological activity, similar to that of Peginterferon alfa-2a.
Our PK study of AL-624 showed that the 4-Gly fraction has a 6-fold increase of AUC and an 11-fold increase of T1/2 compared with daily CIFN. This result suggests that the drug exposure of 1 dose of AL-624 4-Gly might be close to that of 7 daily dosings of CIFN. This prediction was validated in the subsequent YFV hamster study. Similar to HCV, YFV is a positive-strand RNA virus and thus this is a relevant and convenient model for HCV in small animals (Julander and others 2007). Unlike other rodent species, human Type I interferon can interact with hamster Type I interferon receptor and, consequently, activate the innate immune response, resulting in an antiviral effect. The YFV hamster model is a lethal model, and the primary end point is the survival rate after drug treatment. Both AL-624 4-Gly and Peginterferon alfa-2a were dosed at once a week and achieved the same efficacy. Weekly dosing of AL-624 4-Gly also achieved a similar effect as daily dosing of CIFN in terms of survival rate. The secondary end point (serum ALT level) also supported the conclusion that once-a-week dosing of AL-624 4-Gly achieves a similar effect in reducing liver damage as daily CIFN or weekly Peginterferon alfa-2a. Taken together, these results clearly demonstrate that when highly glycosylated, addition of 4 glycosylation sites could improve the PK profile of CIFN to achieve once-a-week dosing.
Once-a-week dosing CIFN, with biological activity similar to that of current market leader Peginterferon alfa-2a, cannot be a viable development candidate. It is our goal to develop a best-in-class molecule in the Type I interferon market. We hope to develop a CIFN variant molecule with even less frequent dosing than Peginterferon alfa-2a while maintaining the high biological activity of its parent. Once-a-month dosing is our ultimate goal. This will further reduce the frequency of injection from the current once per week, cut medical cost, and lighten the burden of treatment on a patient's daily life. It also offers the opportunity to have a nurse administer the injections in a doctor's office during the monthly visit, thus improving the compliance and potential results of treatment.
Design and generate once a month dosing CIFN
Albuferon was the most advanced long-acting IFN to target once-a-month dosing until the recent development suspension (Nelson and others 2009). It is a recombinant protein drug consisting of genetically linked human albumin and IFN-α, and the MW of the total protein is 85.7 kD. The loss of biological activity of Albuferon is more than 20-fold when compared with parent IFN-α (Osborn and others 2002). Human Albumin has unique binding and distribution properties that may play a role in some of the adverse effects for Albuferon in clinical trials. We believe sugar chains display drastically different physical properties compared with Albumin and anticipate that we can avoid some of the adverse effects of Albuferon by using the sugar chains to achieve the gain in MW. To target a total MW of about 90–100 kD, 15 highly glycosylated sugar chains may be needed for CIFN, given the formula of roughly 5 kD per sugar chain. This presents a daunting task for traditional glycosylation methods considering the loss of biological activities due to steric effect. From our CIFN study, it was shown that the addition of 4 internal glycosylation sites can cause a significant reduction in the biological activity.
A plausible way to address the issue is to attach the glycosylation sites to the terminals of the protein. It has been reported that addition of 2 glycosylation sites to the N-terminal of follicle-stimulating hormone increases the T1/2 3- to 4-fold for the glycoprotein (Klein and others 2003). Even for terminal extension, adding 15 sites can be very different from adding just 2 sites. The addition to the N-terminal of CIFN was prioritized because of the reported higher glycosyloation efficiency at the N-terminal of the protein (Bano-Polo and others 2011). We started with a smaller number of site additions and gradually worked our way toward 15 sites, optimizing the site and linker sequences in the process (data not shown). The eventual prototype molecule AL-683 is shown in Fig. 5A: with NIT as the core N-link glycosylation site sequence and GGGV as the linker sequence. Isoleucine (I) was selected to be in the core glycosylation site due to reported strong glycosylation efficiency in its presence (Shakin-Eshleman and others 1996). The sequence of NITGGGV was repeated 14 times to generate 14 external glycosylation sites at the N-terminal of CIFN, while 1 internal glycosylation site was created at position 78. The retention of a limited number of internal glycosylation sites could help reduce the potential protease degradation of the protein. The average size of the expressed products can reach to that of Albuferon, while the biological activity is essentially the same as the unmodified CIFN. These results demonstrate that the addition of glycosylation sites to the N-terminal of CIFN can be a viable approach to drastically increase the MW while maintaining the biological activity. The reason behind the phenomenon is possibly due to the lower steric effects from multiple sugar chains when they are removed from the internal domains of proteins to an external, flexible arm.
Dissimilar to testing once-a-week dosing of AL-624, the small rodent efficacy model to test once-a-month IFN does not exist; the study duration of the YFV hamster model is shorter than 1 month. CHC primate models are prohibitively expensive and difficult to obtain. We decided to study the PK as well as PD responses in the mouse as an alternative. A mouse homologue of AL-683 was generated with mouse IFN-α-1 to carry out mouse PK/PD studies of this type of molecule with external glycosylation site extension. Mouse IFN was chosen, because human IFN cannot induce PD reactions in the mouse due to species differences. The N-terminal sequence addition is identical in AL-683 and its mouse homologue. With its own natural glycosylation site at position 78, the total number of glycosylation sites remains akin to AL-683. Escherichia coli expressed mouse IFN-α-1 was used as an unglycosylated control. The MW of the mouse homologue is around 100 kD and has a far superior PK profile when compared with its unglycosylated parent: a ∼37-fold improvement in T1/2 and ∼33-fold improvement in AUC. The fold of PK improvement compared with the unglycosylated parent far exceeds that of AL-624, the weekly dosed CIFN. This experiment demonstrated the usefulness of our core glycosylation technology in generating hyperglycosylated IFN with a superb PK profile. However, there are limitations in models, exhibited here in extrapolating the mouse homolog data into that of AL-683. In the process of further developing AL-683 into a drug for twice-a-month dosing, and possibly once-a-month dosing, more studies with AL-683 are warranted.
The improvement in PD marker AUC is only a few fold, not proportional to the improvement in PK. A similar observation was found in a PD comparison study with IFN-β-1a and PEGylated human IFN-β-1a in monkeys by Biogen (Pepinsky and others 2001), in which the Neopterin response was not altered with administration of long-acting PEGylated human IFN-β-1a. Little is known about the effect of continuously high IFN in the blood on the IFN pathway. It has been reported that in cultured cells, the IFN signaling becomes refractory under the continuous presence of IFN (Sarasin-Filipowicz and others 2009). However, longer-lasting Peginterferon alfa-2a and Peginterferon alfa-2b clearly provided better sustained virologic response in treating HCV patients when compared with the nonpegylated parents (Shepherd and others 2004).
In conclusion, we have designed and generated a prototype CIFN with 15 glycosylation sites that can be tested for twice-a-month or once-a-month dosing. The biological activity of this prototype remains the same as CIFN, the highest among all of the Type I IFNs. After its successful development, in addition to other benefits mentioned, it is conceivable that the cost structure of such a potent, less frequently administered medicine will be very favorable to the patients. However, many challenges remain in process development and manufacturing to optimize the conditions toward the high degree of glycosylation and the batch-to-batch glycosylation consistency of this prototype in order for it to become more drug like. During the course of our study, we have set up internal glycosylation quality controls (eg, sialic acid content) for the quality of the glycoprotein before PK, PD, and animal efficacy studies. These quality controls will need to be strengthened in the future process development of this prototype.
Furthermore, the technology described here has good adaptability toward many other proteins and good scalability on the number of sugar chains to be incorporated. It could potentially be applied as platform technology, beyond interferons, toward other small protein therapeutics to enhance their PK profile based on their own clinical needs.
Footnotes
Acknowledgments
The authors would like to thank Dr. Biswa Choudhury and his team at Glycotechnology Core Resource, UCSD for providing numerous glycoanalysis tools, including DMB-Sialic Acid assay, MALDI-TOF analysis, and so on.
Author Disclosure Statement
The following authors are current or former employees of Alios BioPharma: Joshua S. Taylor, Qingling Zhang, Antitsa D. Stoycheva, Hua Tan, Christabel V. Moy, Sushmita Chanda, Julian A. Symons, Leo N. Beigelman, Lawrence M. Blatt, and Jin Hong. The following author has no competing financial interest: Justin G. Julander.