
Abstract
Interleukin-17 (IL-17) has been proved to be involved in the pathogenesis of several autoimmune diseases, including lupus, rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease. The regulation of IL-17 signal transduction is less studied. miR-30a has been identified to be downregulated in these human autoimmune diseases and their related animal models. However, how it functions in IL-17-mediated inflammation and the pathogenesis of these diseases remain unknown. In this study, we showed that miR-30a inhibits IL-17-mediated NF-κB and MAPK activation, leading to the reduced production of inflammatory cytokines and chemokines. miR-30a also reduced mRNA stability triggered by IL-17 stimulation. These suppressive effects of miR-30a were mediated by directly targeting Traf3ip2 mRNA (coding for Act1). Thus, we concluded that the downregulation of miR-30a in autoimmune diseases may exacerbate IL-17-mediated inflammation, which may serve as a potential target for the therapy of these diseases.
Introduction
A
IL-17 is the prototype of the IL-17 cytokine family and also the signature cytokine secreted by Th17 cells (Kolls and Linden 2004; Korn and others 2009). IL-17 plays critical roles in host defense against bacterial and fungal invasion, as well as in the pathogenesis of allergy, autoimmune diseases, obesity, and tumors (Song and Qian 2013). We have also identified a pathogenic role of IL-17 in NK cell-mediated acute hepatitis (He and others 2013). The IL-17 signal transduction has only been identified recently (Qian and others 2007; Gaffen 2009). Upon its interaction with its receptors IL-17RA-IL-17RC, adaptor protein Act1 is recruited to the receptors, and then functions as an E3 ligase to ubiquitinate TRAF6. K63-ubiqutinated TRAF6 then activates MAPKs and NF-κB, and finally triggers transcription of several inflammatory cytokines (Song and Qian 2013). Another important biological effect of IL-17 is its ability to maintain mRNA stability. The IL-17-mediated mRNA stability requires Act1, but not TRAF6, indicating the existence of other signal pathways (Hartupee and others 2009). Later studies revealed an involvement of TRAF2 and TRAF5 associated with Act1, which is responsible for mRNA stability (Sun and others 2011).
Since IL-17 has a broad role in the triggering of inflammatory responses, the precise regulation of IL-17 signal is needed to avoid excessive inflammatory injury. TRAF3 was the first identified negative regulator of the IL-17 signal pathway (Zhu and others 2010). TRAF3 competes with Act1 to bind with IL-17R and attenuates both MAPK and NF-κB pathways. TRAF4 is also a negative regulator of the IL-17 pathway by competing with TRAF6 to bind with Act1 (Zepp and others 2012). A deubiquitinating enzyme, the ubiquitin-specific protease USP25, has been recently identified to remove IL-17-induced ubiquitination of TRAF5 and TRAF6 and it consequently suppressed both TRAF6-dependent NF-κB activation and TRAF5-dependent mRNA stabilization (Zhong and others 2012). However, the regulation of the IL-17 signal pathway remains rare.
MicroRNAs are common regulators of gene expression at the mRNA level (Bartel 2004). Its role in the regulation of immune reactions and inflammatory response has also been precisely identified (Taganov and others 2007; O'Connell and others 2010). Recently, Zhu and others (2012) have reported miR-23b in the negative regulation of IL-17-associated autoimmune inflammation by targeting TAB2, TAB3, and IKK-α. However, besides miR-23b, they also identified miR-30a to be obviously downregulated in inflammatory lesions of humans with lupus, rheumatoid arthritis, or multiple sclerosis, as well as in the mouse models with these diseases (Zhu and others 2012). The action and function of miR-30a in IL-17-associated inflammation have not been elucidated.
In this study, we studied the regulatory function of miR-30a in IL-17 signal transduction. miR-30a could negatively regulate IL-17-mediated NF-κB and MAPK activation, and finally reduced the production of inflammatory cytokines and chemokines. In addition, miR-30a also suppressed the mRNA stabilization ability of IL-17. We further proved that miR-30a negatively regulates IL-17 signaling through targeting the adaptor Act1. Overexpression of Act1 abrogated the suppressive effect of miR-30a. Thus, we conclude that in inflammatory lesion of several autoimmune diseases, the downregulation of miR-30a may help exacerbate the inflammatory effect of IL-17, which contributes to the pathogenesis of these autoimmune diseases.
Materials and Methods
Cell lines and reagents
The human cell lines, HeLa and THP1, were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium and RPMI1640 containing 10% fetal bovine serum (Invitrogen). THP1 cells were differentiated into macrophage phenotype by incubation with 50 ng/mL PMA for 48 h. Cell morphology was observed by a phase-contrast microscope, and macrophage surface markers were confirmed by FACS. MicroRNA mimics and inhibitors were purchased from Genepharma. Antibodies to p-ERK, p-JNK, p-p38, p-p65, TRAF6, Act1, and actin were purchased from Santa Cruz Biotechnology, Inc. Actinomycin D was purchased from Sigma-Aldrich.
RNA preparation, reverse transcription, and quantitative real-time PCR
Total RNA was prepared from cultured cells using TriZol Reagent (Invitrogen) according to the manufacturer's instruction. Total RNA was reverse transcribed with random 6-mer primers and oligo dT (18 mers) (Takara). The expression of the genes encoding IL-6, CCL20, and CXCL1 was quantified by real-time polymerase chain reaction (PCR) with the SYBR Premix ExTaq kit (Roche). β-Actin was used as an endogenous control. A 2−ΔΔCt method with StepOne Software V2.1 (AB) was used to calculate fold change. The half-life of mRNA (t 1/2) was calculated as previously described (Zhu and others 2010); briefly, the mRNA level for the gene of interest was plotted as log of the percentage of remaining mRNA versus time. The best fit to linear decay was determined, and the t 1/2 was calculated from the intersection at the point corresponding to 50% residual RNA.
Cell transfection
Cells were transfected with miRNA mimics or inhibitor using INTERFERin (Polyplus transfection) according to the standard protocol. The sequences of miR-30a were as follows: sense, 5′-UGUAAACAUCCUCGACUGG AAG-3′, and antisense, 5′-CUUCCAGUCGAGGAUGUUUACA-3′; the sequence of the 2′-O-methyl-modified miR-30a inhibitor was as follows: 5′-CUUCCAGUC GAGGAUGUUUACA-3′. The sequences of ctrl mimics were as follows: sense, 5′-UUCUCCGAACGUGUCACGU-3′, and antisense, 5′-ACGUGACACGUUCGGAGAA-3′; the sequence of the ctrl inhibitor was as follows: 5′-CAGUACUUUUGUGUAGUACAA-3′ (Genepharma). Transfection of Act1-encoding plasmid was performed using jetPEI transfection reagent (Polyplus).
Luciferase reporter assay
For luciferase reporter assays, wild-type 3′ UTR of Trif3ip2 from human cDNA was cloned into pGL3-promotor vector (Promega). A three-point mutation in the seed sequence was synthesized with a QuikChange site-directed mutagenesis kit (Agilent Technologies). These report vectors were cotransfected with Renilla vector pRL-TK into HeLa cells, and the luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega).
Immunoblot analysis
Cells or mouse tissues were directly lysed in Triton buffer (20 mM Hepes and 0.5% Triton X-100, pH 7.6) and separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Immunoblot analysis was performed by initial transfer of proteins onto polyvinylidene fluoride filters using Mini Trans-Blot (Bio-Rad Laboratories) and followed by a blocking step using Tris-buffered saline with 0.1% Tween 20 plus 5% nonfat dried milk for 1 h at room temperature. The filters were then incubated with primary antibody overnight and subsequently washed. The blots were then incubated with a secondary antibody conjugated with horseradish peroxidase (HRP) for 1 h at room temperature. After extensive washing of the blots, signals were visualized with chemiluminescent HRP substrate (Millipore). Sometimes, the same blot was stripped off and reprobed with other antibodies to check their proteins.
Detection of cytokine production
IL-6, CXCL1, and CCL20 production in the supernatants was measured with ELISA Kits under the manufacturer's instructions (R&D).
Statistical analysis
Data are expressed as mean±SD of experiments. Statistical comparisons were performed using Student's t-test.
Results
miR-30a reduced IL-17-stimulated expression of cytokines and chemokines
Screening of miRNAs differentially expressed in inflammatory tissue from human autoimmune diseases and the related mouse models have shown that miR-30a is downregulated in autoimmune tissues (Zhu and others 2012); however, its role in these diseases remains unknown. To test whether miR-30a has any function in IL-17-mediated signaling, we first transfected miR-30a mimics in to HeLa cells. Interestingly, IL-17-induced inflammatory gene transcription (Il6, Ccl20, and Cxcl1) was reduced by miR-30a mimic transfection (Fig. 1A). These inflammatory cytokines and chemokines were also detected to be reduced in the supernatants by ELISA assay (Fig. 1B). To evaluate the applicability of our finding, we further tested the function of miR-30a mimics in human macrophages derived from THP1 cells. miR-30a indeed suppressed IL-17-induced cytokine production in human macrophages (Supplementary Fig. S1A, B; Supplementary Data are available online at

miR-30a inhibits interleukin-17 (IL-17)-induced inflammatory gene expression.
To confirm this, we also transfected HeLa cells with miR-30a inhibitors and observed IL-17-induced gene expression. In contrast, miR-30a inhibitors increased IL-17-induced Il6, ccl20, and Cxcl1 transcription and also protein secretion in the supernatant (Fig. 1C, D). These have also been confirmed in THP1-derived human macrophages (data not shown). Furthermore, miR-30a inhibited IL-17-induced cytokine transcription in a dose-dependent manner (Supplementary Fig. S2A) and functioned early from 1 h post IL-17 stimulation (Supplementary Fig. S2C). miR-30a inhibitor also functioned in a time and dose-dependent manner (Supplementary Fig. S2B, D). Thus, we concluded that miR-30a has a negative role in IL-17-mediated cytokine production. On this occasion, the reduced expression of miR-30a in autoimmune tissues may prolong IL-17-mediated inflammation, which then exacerbates autoimmune injury.
miR-30a inhibits IL-17-induced MAPK and NF-κB activation
The downstream signaling of IL-17 was proved to activate the NF-κB and MAPK pathways, thus the suppressive effect of miR-30a may be mediated by blocking one of these pathways. To test which pathway miR-30a interferes with, we analyzed the activation of p65, erk, p38, and jun in HeLa cells stimulated with IL-17 at indicated times. miR-30a reduced the phosphorylation of p65, erk, jnk, and p38 after IL-17 stimulation compared with control RNA transfection (Fig. 2A). Accordingly, miR-30a inhibitors increased the activation of both NF-κB and MAPK pathways after IL-17 stimulation (Fig. 2B). These results have also been confirmed in human macrophages derived from THP1 cells (Supplementary Fig. S1C). Thus, the target molecules of miR-30a in IL-17 signal transduction may exist upstream of NF-κB and MAPK pathways. IL-17 signaling initiates from binding to IL-17RA and IL-17RC heterodimeric receptors, and then Act1 is recruited to the receptors and functions as an E3 ubiquitin ligase to polyubiquitinate TRAF6, which then induced the activation of NF-κB and MAPK (Song and Qian 2013). So, the upstream molecules IL-17RA, IL-17RC, Act1, and TRAF6 complex seem to be the potential target of miR-30a.

miR-30a alleviates IL-17-induced signal transduction. HeLa cells transfected with miR-30a mimics
miR-30a reduced the effect of IL-17 on chemokine mRNA stabilization
Besides activation of NF-κB and MAPK pathways, IL-17 can also post-transcriptionally stabilize mRNAs induced by TNF. To test whether miR-30a also influences the mRNA stabilization effect of IL-17, we first stimulated HeLa cells with TNF for 0.5 h to promote the inflammatory gene transcription, and then treated them with IL-17 along with actinomycin D to block transcription for 0.5–3 h. Although CXCL1 mRNA was induced to a similar extent in control and miR-30a-transfected cells after the initial TNF treatment, these mRNAs decayed more rapidly in the miR-30a group (Fig. 3A, B). These results indicated that miR-30a also reduced IL-17-mediated stabilization of CXCL1 mRNAs, which is in accordance with the above hypothesis that miR-30a acts on the most upstream signal molecules.
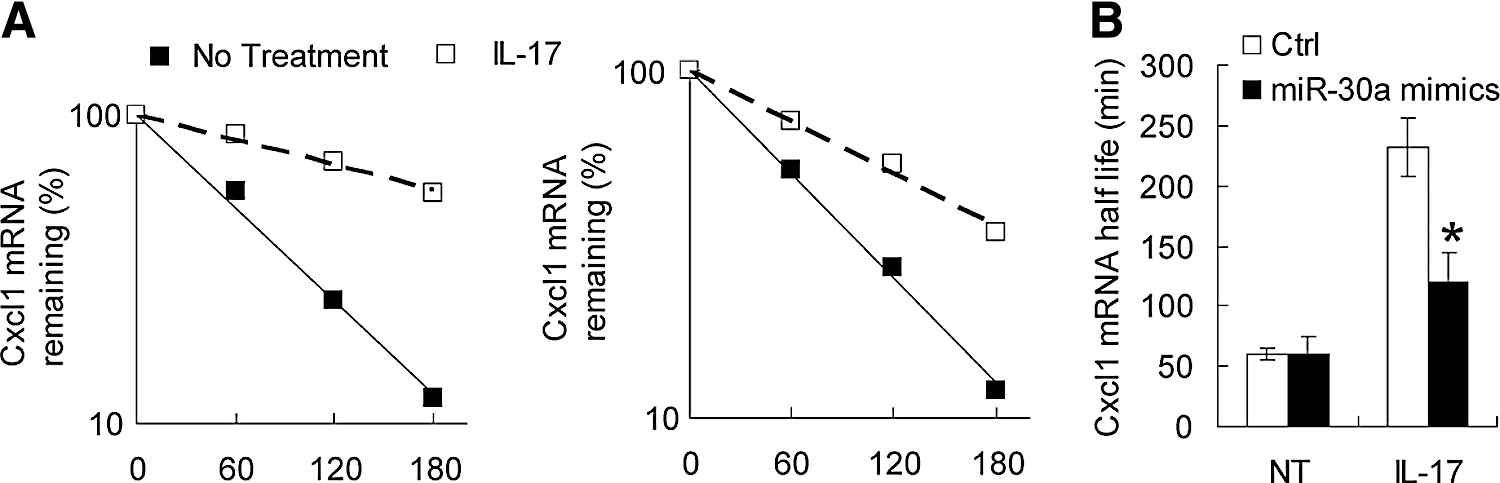
miR-30a reduces IL-17-mediated mRNA stabilization. HeLa cells infected with control (left) or miR-30a (right) mimics, which were pretreated for 2 h with tumor necrosis factor (TNF) (10 ng/mL), followed by actinomycin D (5 mg/mL) alone (no treatment) or in combination with IL-17 (50 ng/mL) for the indicated time. Cxcl1 mRNA was determined by real-time PCR analysis. The results are presented as decay over time
miR-30a targets Act1 downstream of IL-17 signaling
To identify the exact mRNAs that are regulated by miR-30a in IL-17 signaling, we performed a bioinformatics search for putative miRNAs predicted to be targeted by miR-30a using TargetScan and PicTar. Most surprisingly, we found that the 3′ UTR of Act1 mRNA bears the miR-30a-binding sites and these sites are poorly conserved in mammals (Fig. 4A). Meanwhile, Il6, ccl20, and Cxcl1 mRNA were not directly included in the predicted miR-30a targets, suggesting that these cytokines and chemokines induced by IL-17 stimulation were not directly targeted by miR-30a.

miR-30a directly targets Traf3ip2 transcripts.
A dual-luciferase reporter assay was used to confirm the possibility that Act1 was regulated post-transcriptionally by miR-30a. A fragment containing the target site of miR-30a from Act1 3′ UTR was inserted into the luciferase plasmid, pGL3. The expression of luciferase was greatly reduced when the reporter plasmid containing the Act1 3′ UTR was cotransfected with miR-30a mimics, while cotransfection of HeLa cells with mock luciferase plasmids and the miR-30a mimics did not significantly influence luciferase activity (Fig. 4B). Furthermore, this decrease was also abrogated by transfection of the plasmid containing a 3-base mutation in the miR-30a-binding site (Fig. 4B). These data indicate that miR-30a can suppress Act1 expression by degradation of its mRNA or inhibition of its translation.
miR-30 suppresses IL-17 signaling through targeting Act1
To confirm that miR-30a targets Act1, we detected both the mRNA level and protein level of Act1 in HeLa cells after miR-30a mimic transfection. The mRNA of Act1 was reduced during the whole experiments (Fig. 5A), and Western blot analysis showed a more obvious reduction of Act1 expression in the miR-30a mimic group (Fig. 5B). The same data were also confirmed in THP1-derived macrophages (Supplementary Fig. S3). To elucidate whether miR-30a-mediated downregulation of Act1 was responsible for the reduced cytokine and chemokine expression, we overexpressed Act1 mRNA in HeLa cells without 3′ UTR to avoid targeting by miR-30a. As shown in Fig. 5C, overexpression of Act1 greatly rescued the reduction of Il6 and Ccl20 expression by miR-30a. These data indicate that miR-30a-mediated inhibition of IL-17-mediated inflammation was mainly caused by its suppression of Act1 expression.

miR-30a suppresses IL-17 function by targeting Act1.
Discussion
The proinflammatory cytokine IL-17 and Th17 cells have recently been proved to be associated with the pathogenesis of several autoimmune diseases (Korn and others 2009; Segal 2010; Dong and Zhu 2012; Maddur and others 2012). The signal transduction of IL-17 has also emerged in these years. It contains mainly 2 pathways, the TRAF6-dependent and TRAF6-independent pathways (Levin 2009; Gu and others 2013; Song and Qian 2013). The TRAF6-dependent pathway was responsible for the IL-17-induced production of inflammatory cytokines and chemokines, while the TRAF6-independent pathway was mainly mediated by TRAF2 and TRAF5, and contributes to the mRNA stabilization by IL-17. Some studies suggested that TRAF3, TRAF4, and USP25 are negative regulators of IL-17 signaling (Sun and others 2011; Zepp and others 2012; Zhong and others 2012). A microRNA profile has found miR-23b and miR-30a to be downregulated in the inflammatory lesion of autoimmune diseases (Zhu and others 2012). They demonstrated that the reduced miR-23b may contribute to exacerbate IL-17-mediated inflammation by targeting TAB1 and TAB2 in the IL-17 signal pathway. We here proved that reduced miR-30a also exacerbates IL-17-mediated inflammatory injury by targeting the adaptor of IL-17 signaling, Act1.
miR-30a is a member of the miR-30 miRNA family conserved between mammals, which is abundantly expressed in the heart under physiological condition (Small and others 2010). Reduced expression of miR-30a has been reported in a wide range of tumors, including colorectal cancer, thyroid anaplastic carcinomas, nonsmall cell lung cancer, lymphocytic leukemia, and correlated with tumor progression (Calin and others 2004; Cummins and others 2006; Visone and others 2007; Kumarswamy and others 2012; Zhao and others 2014). miR-30a is also able to modulate autophagy through inhibiting beclin 1 expression (Zhu and others 2009). Its role in the immune system remains largely unknown. miR-30 was reported to be downregulated in the liver after hepatitis C virus infection (Pedersen and others 2007), implicating its function in the antiviral effect. However, transfection of miR-30 did not substantially attenuate viral replication (Pedersen and others 2007). CD3 stimulation of T cell also reduced miR-30a expression (Cobb and others 2006), but the activity of miR-30a in T-cell activation is not elucidated. The downregulation of miR-30a has also been reported in several autoimmune diseases (Zhu and others 2012); we proved that this reduced miR-30a may exacerbate IL-17-mediated inflammatory responses by targeting adaptor protein Act1 of IL-17 signal transduction. Recently, Liu and others (2013) reported that miR-30a promotes the autoimmune disease (SLE) by targeting Lyn in B cells, which seems contradictory to our data. However, elevated miR-30a was observed in B cells, the antibody-producing cells, and the mediator of SLE, while reduced miR-30a was observed in the inflammatory lesions (Zhu and others 2012), the target organ of SLE. Besides, miR-30a acts positively in BCR signaling in B cells, while in target macrophages or epithelial cells, it acts negatively in IL-17 signal transduction. Thereafter, these data may illustrate that miR-30a could be acting positively or negatively in different cell types and to different signaling.
In sum, we have provided evidence for a previously unknown regulatory mechanism by discovering that miR-30a acts as a negative regulator of IL-17 signaling. miR-30a mainly targets adaptor protein Act1 to reduce its expression and thus attenuates the downstream activation of both TRAF6-dependent and independent pathways of IL-17, leading to reduced production of inflammatory cytokines and chemokines and impaired stability of mRNA. These data may also suggest a disease-promoting role of reduced expression of miR-30a in the inflammatory lesion of several human autoimmune diseases, including lupus, rheumatoid arthritis, and multiple sclerosis. miR-30a may also be a potential target for the therapy of these diseases.
Footnotes
Acknowledgments
This work was supported by 12th Five-Year Significant New Drugs Creation Plan of the Ministry of Science and Technology of China (No. 2011ZX09302-003-03). Support was provided by Zhejiang Provincial Natural Science Foundation of China (LR12H26001), the 12th Five-Year Major Science and Technology Projects of Infectious Disease about AIDS and Viral Hepatitis (2013ZX10003007), and the National Natural Science Foundation of China (81072339).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.