
Abstract
The cytokine granulocyte colony-stimulating factor (G-CSF) binds to its receptor (G-CSFR) to stimulate hematopoietic stem cell mobilization, myelopoiesis, and the production and activation of neutrophils. In response to exercise-induced muscle damage, G-CSF is increased in circulation and G-CSFR has recently been identified in skeletal muscle cells. While G-CSF/G-CSFR activation mediates pro- and anti-inflammatory responses, our understanding of the role and regulation in the muscle is limited. The aim of this study was to investigate, in vitro and in vivo, the role and regulation of G-CSF and G-CSFR in skeletal muscle under conditions of muscle inflammation and damage. First, C2C12 myotubes were treated with lipopolysaccharide (LPS) with and without G-CSF to determine if G-CSF modulates the inflammatory response. Second, the regulation of G-CSF and its receptor was measured following eccentric exercise-induced muscle damage and the expression levels we investigated for redox sensitivity by administering the antioxidant N-acetylcysteine (NAC). LPS stimulation of C2C12 myotubes resulted in increases in G-CSF, interleukin (IL)-6, monocyte chemoattractant protein-1 (MCP-1), and tumor necrosis factor-α (TNFα) messenger RNA (mRNA) and an increase in G-CSF, IL-6, and MCP-1 release from C2C12 myotubes. The addition of G-CSF following LPS stimulation of C2C12 myotubes increased IL-6 mRNA and cytokine release into the media, however it did not affect MCP-1 or TNFα. Following eccentric exercise-induced muscle damage in humans, G-CSF levels were either marginally increased in circulation or remain unaltered in skeletal muscle. Similarly, G-CSFR levels remained unchanged in response to damaging exercise and G-CSF/G-CSFR did not change in response to NAC. Collectively, these findings suggest that G-CSF may cooperate with IL-6 and potentially promote muscle regeneration in vitro, whereas in vivo aseptic inflammation induced by exercise did not change G-CSF and G-CSFR responses. These observations suggest that different models of inflammation produce a different G-CSF response.
Introduction
I
NSAIDs are known for their ability to scavenge reactive oxygen species (ROS) (Fernandes and others 2004; Costa and others 2006; Purpora and others 2013) and reduce macrophage recruitment to the injury site (Schoenfeld 2012a, 2012b). Consequently, NSAIDS may reduce the long-term regeneration of skeletal muscle following injury (Almekinders and Gilbert 1986; Obremsky and others 1994; Mishra and others 1995; Schoenfeld 2012b). To add to the complexity, inflammation may be dependent on the inflammatory stimulus with acute/chronic disease as well as tissue damage known to produce different cytokine responses with dysregulation of the immune response contributing to a wide range of diseases (Moilanen 2014). Therefore, understanding the inflammatory response to different stimuli is fundamental to developing therapeutic agents for inflammatory conditions. Recently, granulocyte colony-stimulating factor (G-CSF) has emerged as a therapeutic agent for skeletal muscle.
The cytokine G-CSF stimulates hematopoietic stem cell mobilization, myelopoiesis, and particularly the production and activation of neutrophils (Roberts 2005). G-CSF exerts its effects by binding to the G-CSF receptor (G-CSFR, encoded by CSF3R) on target cells (Liongue and others 2009). G-CSF has emerged as a potential therapeutic agent to improve muscle regeneration and force recovery following trauma (Liongue and others 2009).
G-CSF administration to rodents enhances muscle strength and recovery following crush injury (Stratos and others 2007) and increases muscle regeneration following a cardiotoxin injection (Naito and others 2009). While the precise molecular mechanisms by which G-CSF exert its effects remain unknown, G-CSF can modulate and synergize with other cytokines. For example, G-CSF treatment protects rodents against lipopolysaccharide (LPS)-induced toxicity, through suppression of tumor necrosis factor-α (TNFα) (Görgen and others 1992). G-CSF also attenuates the monokine release in LPS-treated whole blood ex vivo (Boneberg and others 2000) and is synergistic with stem cell factor, a costimulatory growth factor in G-CSF-mediated hematopoiesis (Duarte and Frank 2000).
Following damage to skeletal muscle, a plethora of pro- and anti-inflammatory cytokines are released into the circulation (Pedersen 2011; Philippou and others 2012) and, therefore, G-CSF may modulate the inflammatory response in skeletal muscle and aid in repair. This hypothesis is supported by observations that human primary muscle cells produce and release G-CSF in response to stretch-induced damage (Peterson and Pizza 2009). However, primary skeletal muscle cultures are routinely contaminated with G-CSF producing fibroblasts (Kawashima and others 1995; Gaster and others 2001). Importantly, the G-CSFR was identified in skeletal muscle C2C12 myoblasts, myotubes, and mature human and rodent skeletal muscle (Wright and others 2014), and an increase in G-CSF from skeletal muscle may suggest that G-CSF acts in an autocrine loop in skeletal muscle. It still remains whether skeletal muscle is directly producing G-CSF or if G-CSF treatment modulates the inflammatory response in skeletal muscle.
Acute inflammation produces ROS that are thought to be beneficial to skeletal muscle adaptations. However, chronically elevated ROS levels are detrimental to skeletal muscle and surrounding healthy tissue (Mason and Wadley 2014) and, therefore, may have negative effects on the adaptations to acute inflammation. For example, human supplementation with a powerful antioxidant and anti-inflammatory agent N-acetylcysteine (NAC) (Uraz and others 2013) inhibits fatigue and blunts muscle force reduction 24–48 h after eccentric exercise; however, long-term muscle function is hampered (Michailidis and others 2013). This suggests that the adaptive long-term changes in skeletal muscle in response to exercise may be dependent on ROS signaling (Powers and others 2010).
A known stimulator of ROS production is G-CSF (Utsumi and others 1992; Yuan and others 2004). Treatment with NAC inhibits G-CSF stimulation of ROS and consequently inhibits proliferation of the G-CSFR expressing murine pro-B cell line (BAF/3) (Zhu and others 2006). Therefore, anti-inflammatory agents such as NAC may negatively influence skeletal muscle adaptations by inhibiting G-CSF-stimulated ROS production. Whether NAC inhibits G-CSF or G-CSFR expression following eccentric damaging exercise remains to be determined.
As the role and regulation of G-CSF and the G-CSFR in skeletal muscle inflammation is poorly understood, the aim of this study was to determine how G-CSF is regulated in skeletal muscle. To this end, we utilized 2 different inflammatory models: (1) LPS treatment of C2C12 myotubes and (2) aseptic inflammation induced by eccentric damaging exercise in human healthy young males. Our first aim was to determine if G-CSF treatment in vitro blunted the inflammatory response induced by LPS in a commonly used C2C12 myotube cell culture model. Our second aim was to determine the effect of eccentric-damaging exercise on G-CSF and G-CSFR regulation, and finally, to establish whether G-CSF and the G-CSFR response to aseptic inflammation induced by eccentric damaging exercise is redox sensitive by investigating the effect of NAC administration.
Materials and Methods
Cell culture
C2C12 myoblasts (ATCC, Manassas, VA) were incubated at 37°C, 5% CO2 in growth media consisting of high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Once cells became confluent and showed signs of spontaneous differentiation, high-glucose DMEM supplemented with 2% horse serum (Invitrogen) replaced the growth media for 5 days. Growth and differentiation media were replaced every 48 h throughout the experiments, unless stated otherwise. At the beginning of the experiment, the cell culture media were removed and replaced with serum-free DMEM containing either the vehicle, 1 μg/mL LPS (E. coli 0111:B4; Sigma-Aldrich, Sydney, Australia) to induce an inflammatory response (Frost and others 2003), G-CSF (100 ng/mL), or the combination of LPS and G-CSF. After 1, 2, 4, and 24 h, cell culture media were removed, immediately snap frozen in liquid nitrogen, and stored at −80°C for cytokine analysis. The cell layer was washed in ice cold phosphate-buffered saline (PBS) and harvested for RNA extraction.
Eccentric exercise and NAC trial
Muscle and plasma samples were obtained from a larger study (Michailidis and others 2013). In brief, 10 healthy, trained, nonsmoking male volunteers participated in this study. Participants were nonsmokers, recreationally trained, and had been engaged in regular exercise at least 3 times per week for more than 12 months. The study comprised of a double-blind, crossover, repeated measures study design, where subjects received either NAC or placebo (PLA) supplementation immediately postexercise and daily for 8 days thereafter (Michailidis and others 2013). Trials were performed in a random order with a 6-week washout period between trials (Michailidis and others 2013). Muscle and blood samples were collected pre-exercise, 2, 48 h, and 8 days postexercise (Michailidis and others 2013). Subjects' characteristics before each trial are shown in Table 1.
BMI, body mass index; NAC, N-acetylcysteine.
Acute leg eccentric exercise was performed on an Isoforce (TUR GmbH, Berlin, Germany) isokinetic dynamometer with participants performing 300 unilateral repetitions consisting of 15 repetitions per set at 30°/s with a 30-s rest between sets (Michailidis and others 2013). This protocol has previously been shown to induce severe myofibrillar disruption and a pronounced inflammatory response (Raastad and others 2010; Michailidis and others 2013).
Immediately postexercise, subjects ingested a NAC or PLA solution orally in 3 daily doses. The solutions consisted of 375 mL water, 125 mL sugar free cordial, and 2 g low-calorie glucose/dextrose powder (Michailidis and others 2013). During the NAC trial an additional 20 mg/kg/day NAC (Uni-Pharma, Cairo, Egypt) was diluted in the solution. Blinded pilot trials confirmed no difference in palatability between the 2 drinks.
The muscle biopsy procedure has previously been described (Michailidis and others 2013). In brief, the initial (baseline) muscle biopsy sample was taken from the middle portion of the vastus lateralis ∼25–30 cm from the midpatella at a depth between 4 and 5 cm after administration of the local anesthetic, xylocaine (1%), using the Bergstrom needle technique with the application of manual suction as previously described (Evans and others 1982). Subsequent biopsy samples were obtained 3 cm proximal to each previous biopsy site. Adipose tissue was removed, and samples were immediately frozen in liquid nitrogen and stored at −80°C. Approximately 20 mg of the muscle samples were homogenized in ice cold extraction buffer (50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% triton-X, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM dithiothreitol, 10 μg/mL trypsin inhibitor, 2 μg/mL aprotinin, 1 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride). The lysate was centrifuged (12,000 g, 20 min, 4°C) and protein concentrations of the supernatants were determined from the development of a standard curve using the bicinchoninic protein assay kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Muscle lysates were diluted to 1 μg/mL for cytokine analysis. Blood samples (15 mL) were drawn from the antecubital fossa using a 20-gauge disposable needle equipped with a Vacutainer tube holder (Becton Dickinson, Franklin Lakes, NJ) in a seated position at 7:00 a.m. following an overnight fast. For plasma separation, a blood portion was collected in tubes with EDTA and centrifuged (1,370 g, 10 min at 4°C).
Quantitative real-time polymerase chain reaction
RNA was isolated using TRI-Reagent® (Ambion, Inc., Austin, TX) according to the manufacturer's protocol. RNA concentration was determined by the absorbance at a wavelength of 260 nm using the NanoDrop® ND-1000 spectrophotometer (NanoDrop Products, Wilmington, DE). RNA was treated with DNAse amplification grade I (Invitrogen) before being reverse transcribed to synthesize the first-strand cDNA using the high-capacity RNA-cDNA reverse transcription kit (Applied Biosystems, Forster City, CA). All cDNA was diluted to a working concentration of ∼5 ng/μL quantitative real-time polymerase chain reaction (qRT-PCR) was used to measure the messenger RNA (mRNA) expression. The primers for the genes of interest, G-CSF, interleukin (IL)-4, IL-6, IL-10, IL-12, IL-18, monocyte chemoattractant protein-1 (MCP-1), and TNFα were designed using the web-based software Primer 3 (
IL, interleukin; MCP-1, monocyte chemoattractant protein-1; TNFα, tumor necrosis factor-α.
Western blotting
Electrophoresis was performed using a 4%–12% NuPAGE® Novex Bis-Tris Gel in NuPAGE SDS MOPS Running Buffer (Invitrogen). Proteins were transferred to a PVDF membrane (Millipore, North Ryde, Australia) in a Bjerrum buffer containing 50 mM Tris, 17 mM glycine, and 10% methanol. The membranes were blocked with 5% bovine serum albumin (BSA) in PBS, after which they were incubated overnight at 4°C with anti-G-CSFR antibody (Santa Cruz, Dallas, TX) diluted in 5% BSA in PBS. Following washing, the membranes were incubated for 1 h with goat anti-rabbit IgG antibody (1:5,000) labeled with an infrared-fluorescent 800 nm dye (Alexa Fluor® 800; Invitrogen) in PBS containing 50% Odyssey® blocking buffer (LI-COR Biosciences, Lincoln, NE) and 0.01% sodium dodecyl sulfate (SDS). Proteins were exposed on an Odyssey Infrared Imaging System (LI-COR Biosciences) and densitometry was achieved using Odyssey Application Software 3.0 (LI-COR Biosciences). GAPDH (1:5,000) (G8795; Sigma-Aldrich) and rabbit anti-mouse IgG antibody labeled with an infrared-fluorescent 680 nm dye (Alexa Fluor® 680; Invitrogen) was used as a loading control.
Cytokine analysis
A Milliplex assay (Millipore) was used to analyze the cytokine expression in the cell culture media, plasma, and muscle lysates. The cytokine detection kits were designed for the simultaneous analysis of multiple cytokines, G-CSF, TNFα, IL-6, and MCP-1, for the cell culture experiments and a singleplex for G-CSF for plasma and muscle lysates. The assays were conducted following the manufacturer's protocol (Millipore, Billerica, MA). Briefly, a standard curve with a range of 10,000–0.64 pg/mL was generated from the supplied cytokine standard. Then, 25 μL of samples and standards were added to a 96-well plate containing the premixed beads coated with the target antibody. Following an overnight incubation, plates were washed twice in wash buffer, and 25 μL of premixed detection antibodies were added and allowed to incubate. After 60 min, 25 μL of streptavidin–phycoerythrin was added to each well and allowed to incubate for a further 30 min. After incubating, wells were washed 2 times in wash buffer and resuspended in 150 μL of sheath fluid. The plate was then analyzed on the Bio-Plex Suspension Array System (V.5.0; Bio-Rad, Hercules, CA) using the 5-PL curve. Samples were run in duplicate, using the low sensitivity (low PMT) function. For the cell culture media intra-assay coefficient of variations (CV%) were as follows, G-CSF: 4.7%, IL-6: 3.5%, and MCP-1: 5.5%. For the NAC trial CV% were, Plasma G-CSF 3.2% and muscle lysates G-CSF 7.9%.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4.1 (GraphPad Software, San Diego, CA). For the in vitro cell culture experiments means were compared using 1-way repeated measures analysis of variance (ANOVA) comparing treatment for each time point. Any significant differences were analyzed using a Newman–Keuls post hoc test. Data are presented as mean±standard error of the mean. Significance was set at P<0.05.
For the human exercise trial, changes in dependent variables were examined with a 2-factor, repeated measures ANOVA [trial(PLA and NAC)×time (pre, 2 h post, 2 days post, 48 h postexercise) using SPSS version 21 (SPSS, Inc., Chicago, IL). Significance was set at P<0.05. When a significant interaction was detected, post hoc analysis was performed through the Bonferonni adjustment.
Results
Cell culture mRNA expression
LPS significantly increased G-CSF mRNA expression by ∼4- and 17-fold above the control group at 1 and 2 h, respectively, while returning to baseline at 4 and 24 h. LPS treatment significantly increased IL-6, MCP-1, and TNFα mRNA expression (Fig. 1), but had no effect on the mRNA expression of IL-4, IL-10, IL-12B, or IL-18 (Fig. 2).
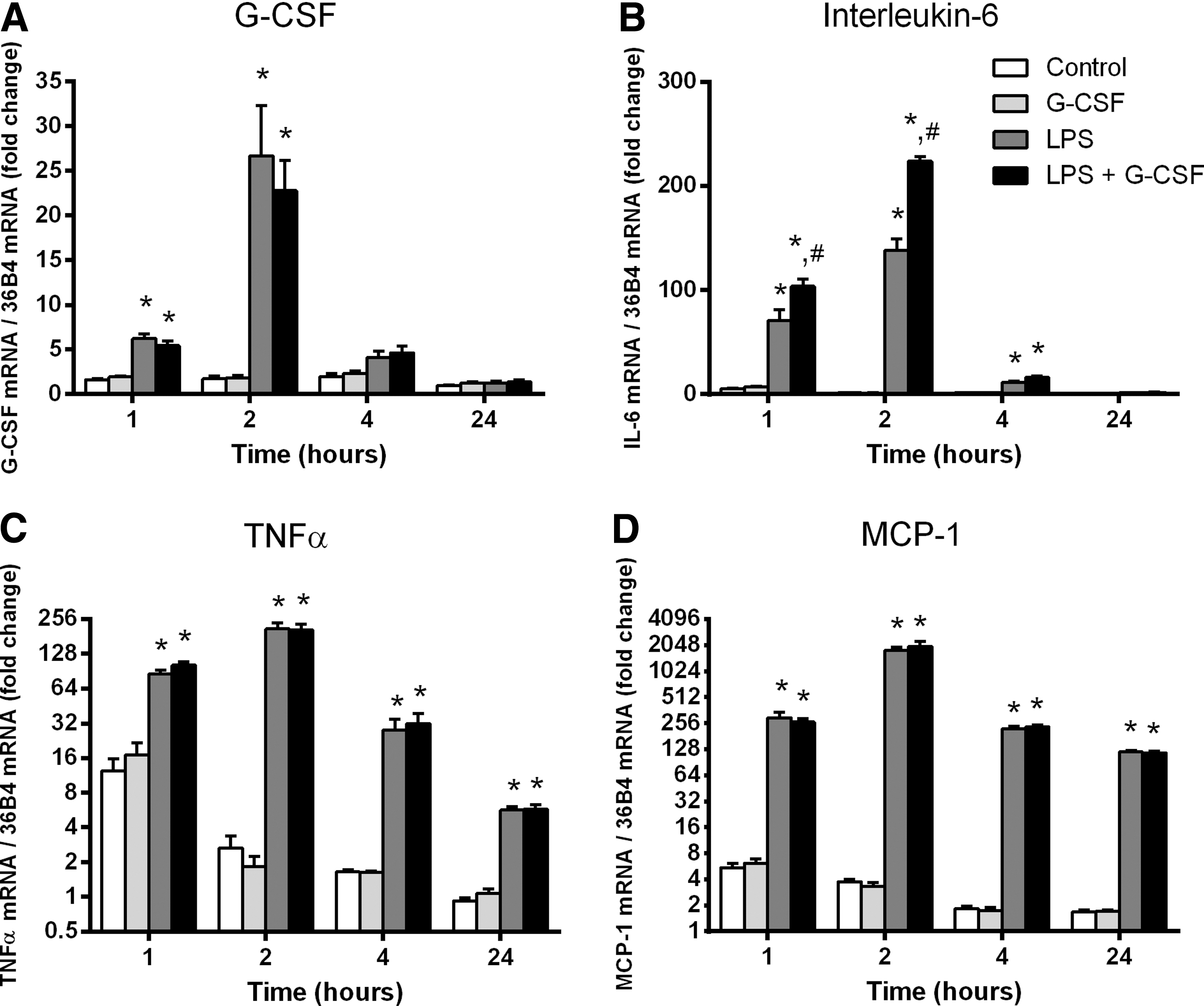
Messenger RNA (mRNA) expression of granulocyte colony-stimulating factor (G-CSF), tumor necrosis factor-α (TNFα), interleukin (IL)-6, and monocyte chemoattractant protein-1 (MCP-1) in C2C12 myotubes following lipopolysaccharide (LPS) and G-CSF stimulation.

mRNA expression of IL-4, IL-10, IL-12β, and IL-18 in C2C12 myotubes following LPS and G-CSF stimulation.
To determine if G-CSF could induce cytokine expression or modulate the inflammatory response, C2C12 myotubes were treated with 100 ng/mL G-CSF in the presence or absence of LPS. Treatment with G-CSF had no effect on G-CSF mRNA expression at any time point (Fig. 1A). G-CSF treatment had no effect on baseline IL-6 mRNA. However, the addition of G-CSF to LPS significantly elevated IL-6 above that of LPS alone, peaking 1.6-fold above the LPS-treated group at 4 h and returning to baseline by 24 h (Fig. 1B). MCP-1 and TNFα were not affected by G-CSF alone nor was MCP-1 or TNFα influenced by G-CSF in the presence of LPS (Fig. 1). Similarly G-CSF treatment had no effect on the expression of IL-4, IL-10, IL-12B, or IL-18 in the presence or absence of LPS (Fig. 2).
Cell culture cytokine analysis
To confirm the production and release of cytokines from C2C12 myotubes treated with LPS, the media were analyzed using a Milliplex assay (Millipore) for the concentration of G-CSF, IL-6, MCP-1, and TNFα. Although G-CSF levels in the media were below the detectable limits (<DL) of the assay in the control group, it was detectable following LPS treatment at 2, 4, and 24 h peaking at 144 pg/mL (Table 3). LPS treatment significantly increased IL-6 at all time points compared to the control group (Table 3). MCP-1, while <DL in the control group was detectable following LPS stimulation after 2 h (730±66 pg/mL) and was significantly greater (P<0.0001) than the control group at 4 h (3,683±63) and 24 h (8,654±172) (Table 3). The concentration of TNFα in the media was not detectable using the cytokine assay at any time point.
Data are mean±SEM, n=6, * P<0.05, *** P<0.001 compared to control at the same time point. † P<0.001 compared to LPS treatment at the same time point. <DL below detectable limits. LPS, 1 μg/mL; G-CSF, 100 ng/mL.
G-CSF, granulocyte colony-stimulating factor; LPS, lipopolysaccharide; SEM, standard error of the mean.
To determine if G-CSF could induce cytokine release from C2C12 myotubes or modulate the inflammatory response induced by LPS, C2C12 myotubes were treated with 100 ng/mL G-CSF in the presence or absence of LPS and the media analyzed for IL-6, MCP-1, and TNFα. G-CSF alone had no effect on IL-6 concentrations, however, at 24 h, the LPS-stimulated IL-6 concentration was significantly higher with the addition of G-CSF; the control group was 6,682±310 pg/mL, while the addition of G-CSF significantly increased the concentration of IL-6 by 1.3-fold to 8,811±328 pg/mL (Table 3). G-CSF had no effect on MCP-1 levels nor did G-CSF modulate the MCP-1 response to LPS (Table 3). Again, the concentration of TNFα was not detectable using the cytokine assay.
Eccentric exercise and NAC supplementation
The eccentric exercise caused skeletal muscle damage resulting in a decrease in mean torque and was sufficient to induce inflammation (Michailidis and others 2013). In particular, following eccentric exercise macrophage accumulation in the muscle increased progressively in both groups; however, blunted macrophage infiltration at 48 h postexercise was observed with NAC treatment (Michailidis and others 2013). NAC treatment also blunted the inflammatory response with lower increases of creatine kinase, C-reactive protein, circulating neutrophils, IL-1β, IL-6, IL-8 coupled with an increase in the IL-10 response (Michailidis and others 2013). Furthermore, recovery with NAC was blunted at 8 days postexercise (Michailidis and others 2013).
Therefore, the aim of this study was to investigate the effects of eccentric exercise-induced muscle damage on G-CSF in plasma and muscle homogenates and to determine if G-CSF is responsive to NAC treatment. A significant main effect for time (P=0.015) for G-CSF plasma levels following skeletal muscle damage was observed. Post hoc analysis revealed a significant elevation in plasma G-CSF 2 h postexercise compared to pre-exercise (Fig. 3). However, there was no main effect for treatment (PLA vs. NAC). G-CSF levels in skeletal muscle did not change with eccentric exercise-induced damage or NAC treatment.
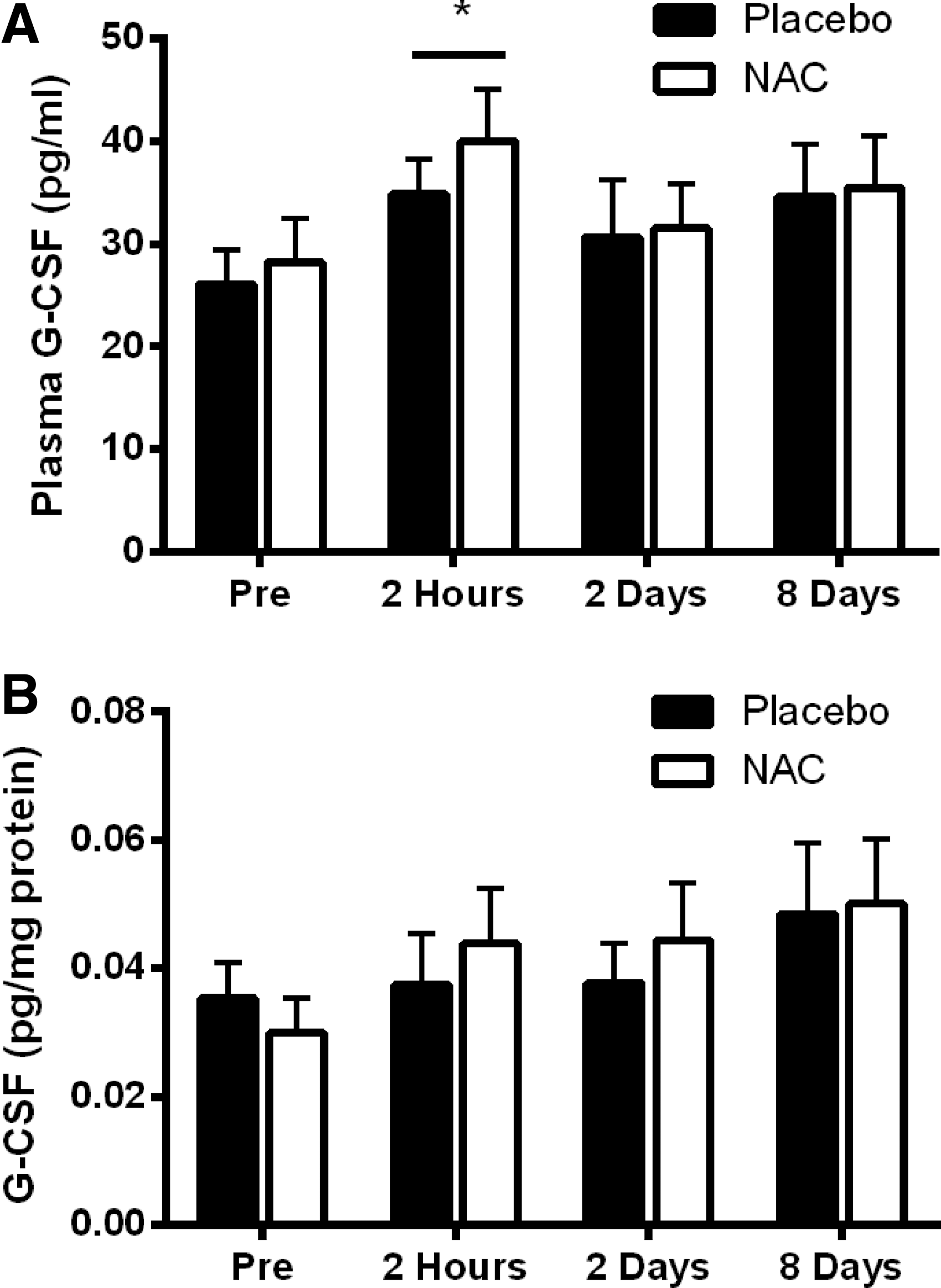
Plasma and skeletal muscle G-CSF levels following eccentric damaging exercise and N-acetylcysteine (NAC) supplementation.
No changes were observed in G-CSFR protein expression in the muscle homogenates following eccentric damaging exercise or NAC supplementation (Fig. 4).

Granulocyte colony-stimulating factor receptor (G-CSFR) expression in skeletal muscle homogenates following eccentric damaging exercise and NAC supplementation.
Discussion
Following skeletal muscle damage G-CSF is increased in circulation (Hirose and others 2004; Paulsen and others 2005; Suzuki and others 2006). Furthermore, skeletal muscle has been identified as a source of cytokine production (Nielsen and Pedersen 2007; Pedersen and others 2007; Pedersen and Febbraio 2008). This suggests that G-CSF may be produced by muscle following an inflammatory stimulus. Therefore, we determined if G-CSF was secreted from muscle cells in vitro using LPS treatment of C2C12 myotubes and if G-CSF was elevated in muscle tissue following exercise-induced aseptic inflammation in humans. Second, it was of interest to determine if G-CSF treatment blunted the inflammatory response in vitro. Finally, we determined if G-CSFR is modulated following eccentric resistance exercise and if the G-CSF/G-CSFR is redox sensitive in vivo. Several novel observations were made. In vitro, LPS stimulated the release of G-CSF from C2C12 myotubes. G-CSF treatment further increased the upregulation of IL-6 when combined with LPS in C2C12 myotubes. In vivo, after eccentric damaging exercise, G-CSF was elevated in the plasma, but not in skeletal muscle tissue. NAC supplementation had no effect on either G-CSF or the G-CSFR levels.
This study shows for the first time that G-CSF is released from myotubes in response to a septic inflammatory stimulus (LPS) which suggests that G-CSF may act as a myokine during inflammation. LPS treatment is used to simulate the effects of sepsis in cell culture models and rodent studies in vivo (Nemzek and others 2008). Sepsis is associated with an increase in skeletal muscle atrophy and inflammation (Callahan and Supinski 2009; Aversa and others 2011; Holecek 2012). During sepsis an early increase in circulating G-CSF is associated with survival (Weiss and others 2001), while G-CSF treatment further increases survival in this model (Görgen and others 1992). The significance of the elevated G-CSF following LPS-induced inflammation remains to be determined; however, these results suggest that the G-CSF response may be specific to septic inflammation.
In a second model of inflammation, circulating G-CSF levels were elevated by 40% after aseptic inflammation induced by damaging exercise, which is in agreement with previous observations (Paulsen and others 2005). Under normal physiological conditions, G-CSF levels are virtually undetectable in the serum (Watari and others 1989). During extreme conditions such as sepsis, G-CSF levels rise to around 0.4 ng/mL (Bozza and others 2007), with some studies reporting patients with G-CSF levels around 3–4 ng/mL (Pauksen and others 1994). Furthermore, recombinant G-CSF can be administered subcutaneously or intravenously with maximal concentrations being reached after 2–8 h with a peak >40 ng/mL (de Haas and others 1994). In our study, plasma G-CSF levels remained in the picogram range, consistent with plasma G-CSF levels following various modalities of exercise (Bonsignore and others 2002; Yamada and others 2002). However, we did not observe any changes in skeletal muscle levels of G-CSF nor did we observe changes with NAC treatment. This suggests that skeletal muscle may not produce G-CSF in response to eccentric resistance exercise and that any increase in circulating G-CSF is a result of the systemic inflammation. Furthermore, this suggests that the G-CSFR response to exercise is not antioxidant dependant. The eccentric resistance exercise protocol increased circulating neutrophils, increased macrophage infiltration, and systemically elevated proinflammatory cytokines in this subject cohort (Michailidis and others 2013). Therefore, the role of G-CSF during aseptic inflammation may be confined to its well-documented role in the mobilization of bone marrow-derived cells.
In this study, LPS stimulation significantly increased IL-6 and TNFα mRNA in C2C12 myotubes, while G-CSF further enhanced LPS-induced IL-6 production. The role of IL-6 in skeletal muscle is not clearly understood. Myoblasts from IL-6 knockout mice display a reduced proliferation rate in vitro (Serrano and others 2008). During exercise IL-6 is consistently increased, and the majority of IL-6 production is suggested to be muscle derived (Pedersen and Fischer 2007). Genetic loss of IL-6 results in a reduced hypertrophic response to functional overloading in rodent muscle (Serrano and others 2008), indicating that IL-6 is required for the hypertrophic responses to resistance exercise, possibly by controlling the inflammatory response.
The increase in IL-6 precedes anti-inflammatory cytokine expression (Pedersen 2007), whereas IL-6 inhibits proinflammatory cytokines such as TNFα during exercise (Starkie and others 2003). An acute rise in IL-6 may be beneficial to muscle growth and hypertrophy, however, chronically elevated IL-6 induces muscle atrophy (Munoz-Canoves and others 2013). Sepsis on the other hand produces a different inflammatory profile by elevating TNFα before the rise in IL-6. This elevation in TNFα/IL-6 in sepsis is considered causal to sepsis-induced protein loss (Kayacan and others 2006).
This response however is rarely seen with exercise-induced aseptic inflammation (Petersen and Pedersen 2005; Petridou and others 2007). G-CSF prevents death by LPS-induced toxicity in rodents (Görgen and others 1992). In rodents, TNFα, IL-6, IL-8, and IL-1ra are elevated when G-CSF is administered immediately before LPS; however, G-CSF administered 24 h before LPS lowers IL-1ra and attenuates IL-8 and IL-6 (Pajkrt and others 1997). In the present study the simultaneous administration of both G-CSF and LPS increased IL-6 by another 1.6-fold above the initial LPS-induced increase. As IL-6 synergizes with G-CSF to promote tumor growth and chemotaxis of bone marrow neutrophils (Yan and others 2013), the increase in vitro from muscle cells may have a physiological relevance in vivo for skeletal muscle.
During sepsis up to 7,500-fold increases in IL-6 are observed (Hack and others 1989; Bossink and others 1995) and while a modest 1.6-fold increase may seem irrelevant, comparisons between media from in vitro models and serum from in vivo models are difficult. Thus, a 1.6-fold increase may have significant effects in vivo suggesting that the protective effects of G-CSF during sepsis, may extend to skeletal muscle through the cooperation of G-CSF/IL-6.
Human primary skeletal myotubes release G-CSF in response to stretch-induced damage in vitro (Peterson and Pizza 2009). However, since G-CSF is a known fibroblast growth factor (Mendoza and others 1990) and fibroblasts may contaminate primary satellite cell populations (Gaster and others 2001), the potential for G-CSF to be released by fibroblasts in primary cell cultures cannot be ruled out. Hence, skeletal muscle may not be the site of G-CSF production during exercise-induced damage. The increase in circulating G-CSF observed 2 h postexercise may be stimulated by an increase in proinflammatory cytokines during exercise-induced inflammation. Eccentric exercise (Hirose and others 2004; Paulsen and others 2005), high-intensity endurance exercise (Suzuki and others 2006), and incremental exhaustive exercise (Suzuki and others 2002) cause significant muscle damage and increase the levels of circulating cytokines, including G-CSF. Therefore, the increase in circulating G-CSF postexercise may simply be systemic and not muscle derived.
Endothelial cells also produce G-CSF (Tura and others 2010). They may release G-CSF directly into the circulation following muscle damage to mobilize bone marrow stem cells and granulocytes or to induce chemotaxis in neutrophils. Thus, an increase in G-CSF would be observed in circulation, but not necessarily within the muscle bed. However, the precise site of G-CSF production during exercise-induced aseptic inflammation remains unknown and muscle-derived G-CSF cannot be ruled out. For example, TNFα mRNA increases markedly in muscle during exercise, but only a small amount of protein is secreted into circulation (Nieman and others 2004). Furthermore, the time points in which the muscle biopsies were taken may not have been optimal to detect changes in G-CSF within the muscle.
The G-CSFR was also measured following eccentric resistance exercise and remained unaltered in skeletal muscle tissue. We have previously shown that the G-CSFR is downregulated in the mdx mouse, a rodent model of Duchenne muscular dystrophy (Wright and others 2014). Therefore, the G-CSF/G-CSFR may be regulated in chronic diseased skeletal muscle, but not in healthy muscle or following aseptic inflammation.
We observed that NAC did not affect elevated circulating G-CSF levels following eccentric damaging exercise. This suggests that G-CSF regulation in muscle in response to damaging exercise is not redox dependent. Previous published data from this sample set showed an incomplete muscle recovery 8 days postexercise in response to NAC supplementation (Michailidis and others 2013). This was associated with a blunted cytokine inflammatory response and a blunted response to redox-sensitive pathways in the NAC trial. NAC is known to inhibit G-CSF production of ROS and lead to reduced proliferation of BAF/3 cells (Zhu and others 2006). Therefore, in the current study, NAC supplementation may have blunted G-CSF-mediated ROS production. However, this remains to be determined.
In conclusion, LPS-induced sepsis elevated G-CSF mRNA expression and G-CSF release in C2C12 myotubes. Although G-CSF treatment did not affect basal levels of TNFα, IL-6, or MCP-1, when G-CSF was combined with LPS, IL-6 mRNA was markedly elevated coinciding with an increase in IL-6 released into the media. These findings suggest that G-CSF may cooperate with IL-6 during sepsis and potentially promote muscle regeneration by mobilizing satellite cells or BMDSC. On the other hand, during damaging exercise-induced aseptic inflammation G-CSF was elevated in circulation at 2 h postexercise; however, there was no increase in the muscle. These results suggest that different models of inflammation may produce a different G-CSF response. The precise role of G-CSF in skeletal muscle adaptation or maladaptation in different models of systemic or local inflammation remains to be determined.
Footnotes
Author Disclosure Statement
No competing financial interests exist.