
Abstract
In this review we discuss the current literature on the effects of type I interferons (IFN) and their downstream effectors on vascular growth in experimental models in vitro and in vivo. In addition to its well-documented role in angiogenesis, that is, the growth of new capillaries from existing vessels, we will also describe emerging evidence and mechanisms by which type I IFN may inhibit arteriogenesis, that is, the expansive remodeling of existing collateral arteries. Crucial in both processes is the common role of circulating monocytes, which are known to act as pivotal cellular modulators in revascularization through secreted chemokines, proteases, and growth factors. These secreted molecules, which are all modulated by IFN signaling, act via degradation of the extracellular matrix and by stimulating the proliferation of vascular smooth muscle cells and endothelial cells. Thus, next to the antiviral and immunomodulatory activities of type I IFNs, a potent role of IFN-β as modulator of revascularization is now emerging and may be considered a potential clinical target for the stimulation of angiogenesis and arteriogenesis in ill-perfused tissues.
Introduction
R
The number of patients suffering from cardiovascular disease is rising globally and myocardial infarction remains the leading cause of death worldwide, identifying sufficient cardiac perfusion as a most critical clinical issue. Several risk factors, including high cholesterol levels, contribute to atherosclerotic obstructive disease and myocardial damage (de Groot and others 2009; Gersh and others 2010). Natural restoration of perfusion can be achieved via the expansive growth of collateral vessels, that is, arteriogenesis, which constitutes a very potent mechanism to increase blood flow. According to Poiseuille's equation a 2-fold increase in diameter will increase blood flow by a factor 16 (vessel radius to the fourth power). A well-developed collateral circulation can alleviate symptoms of chest pain and can be lifesaving in the setting of an acute myocardial infarction (Meier and others 2007). Arteriogenesis involves a complex interplay of several growth promoting and matrix degrading factors (Schaper and Scholz 2003; van Royen and others 2009). Unfortunately, factors that promote arteriogenesis factors have been shown to also promote atherosclerosis, making them unsuited for therapeutic use. Thus, despite the well-recognized need for revascularization therapies, as yet no factor has been identified that efficiently stimulates arteriogenesis in a clinical setting, without unwanted side effects (Schirmer and others 2009).
The neutralization of endogenous interferon (IFN)-β is now emerging as a promising strategy to increase tissue perfusion, because IFN-β inhibits both angiogenesis and arteriogenesis, while it promotes atherosclerosis (Goossens and others 2010).
The present review will therefore focus on the known and potential roles of IFN-β and its downstream effector molecules in vascularization of ischemic tissue.
Interferons
IFNs are glycoproteins that are produced and released by cells of the immune system such as antigen-presenting cells, T cells, and natural killer (NK) cells and also by nonhematopoietic cells, that is, fibroblasts (Schroder and others 2004; Honda and others 2006). The IFN family comprises type I IFN, type II IFN, and type III IFN, which can be further divided in several subtypes. Type I IFNs include the classes IFN-α, IFN-β, IFN-ɛ, IFN-κ, and IFN-ω (Platanias 2005; Kalliolias and Ivashkiv 2010). Type II IFN consists only of IFN-γ and type III IFNs can be divided into IFN-λ1, IFN-λ2, and IFN-λ3 (Kalliolias and Ivashkiv 2010). The main focus of the present review will be on type I IFNs, which are most extensively studied for their role in vascular proliferation.
Type I IFNs are released by immune cells after induction by pathogen-associated microbial patterns (PAMPs) such as viruses, double-stranded RNA, or other microorganisms, or by danger-associated molecular patterns (DAMPs), which are endogenous molecules released from necrotic or dying cells. PAMPs and DAMPs are sensed by Toll-like receptors (TLRs). These TLRs are present on monocytes, macrophages, and dendritic cells that form the first line of defense in innate immunity in mammalian species (Kumar and others 2011). A total of 10 functional TLRs (TLR1-10) have been identified in humans. All TLRs with the exception of TLR5 have been linked to the induction of type I IFNs (Dietrich and others 2010; Kumar and others 2011; Ohto and others 2014). Dietrich and others (2010) showed that heterodimers TLR2-TLR1 and TLR2-TLR6 also induce type I IFN responses. TLR4 is able to induce type I IFN production by recognizing lipopolysaccharide (LPS) from the outer membrane of gram-negative bacteria (Kobayashi and others 2006; Kumar and others 2011). In addition to TLRs, the intracellular recognition receptors retinoic acid inducible gene 1 (RIG1) and melanoma differentiation-associated gene 5 (MDA5), are able to sense intracellular (viral) dsRNA, and induce TLR-independent production of type I IFN (Honda and Taniguchi 2006). In addition to the endosomal-based DNA-sensing receptor TLR9, several other intracellular sensors of dsDNA including DNA-dependent activator of IFN-regulatory factors (DAI, also known as DLM-1 and ZBP1) and STING induce type I IFNs (Yanai and others 2009; Paludan and Bowie 2013). These sensors mediate responsiveness, not only to bacterial, viral, and eukaryotic pathogen-derived DNA but also to host DNA escaped from apoptotic DNA degradation, resulting in the production of type I IFNs (Okabe and others 2005; Yanai and others 2009). Released type I IFNs interact in both a paracrine and an autocrine manner with the type I receptor complex consisting of the IFN-α/β receptors-1 and -2 (IFNAR-1 and -2) leading to the activation of the janus kinase/signal transducers and activators of transcription (STATs), referred to as the JAK/STAT pathway and to activation of additional IFN-regulated transcription factors, which is extensively reviewed elsewhere (Bekisz and others 2004; Platanias 2005; Borden and others 2007; Ivashkiv and Donlin 2014).
For a long time it was believed that IFNs only play a role in interfering with viral replication (Vilcek 2006; Borden and others 2007; Zhang and others 2008b). However, it is now evident that IFNs are not only antiviral cytokines, but also display pleiotropic biologic properties, including antiproliferative, antitumor, and immunomodulatory characteristics. Subsequently, increased IFN production and activity were related to a variety of diseases, including systemic lupus erythematosus (Baechler and others 2003), multiple sclerosis (van Baarsen and others 2006), systemic sclerosis (Bos and others 2009), dermatomyositis (Baechler and others 2007), and rheumatoid arthritis (van der Pouw Kraan and others 2007), and are associated with markers of cardiovascular disease (Somers and others 2012). It appears that type I IFN may play a distinct role in different diseases, with both pro-and anti-inflammatory capacities (Javed and Reder 2006; Kalliolias and Ivashkiv 2010).
Activated TLR4 was shown to be essential in experimental arteriogenesis (Arras and others 1998; de Groot and others 2011). However, unrestrained activation of TLR4 impairs arteriogenesis in the hind limb arterial ligation model (Bastiaansen and others 2014). Likewise, MyD88-dependent signaling is required for angiogenesis, shown in an ex vivo model (Aplin and others 2014), but extensive TLR4 activation inhibits angiogenesis (Huang and others 2013). Schirmer and others (2008) showed that after ex vivo LPS stimulation of TLR4 on patient-derived monocytes, an enhanced IFN-β production and increased type I IFN response program correlates with a poor collateral circulation in patients with coronary occlusions. In vivo, endogenous ligands fulfill a similar role as LPS in activating TLR4 by acting as earlier mentioned “danger signals” (Arslan and others 2010, 2011). These endogenous TLR4 ligands may be extracellular matrix molecules and degradation products thereof or cell-derived stress molecules such as heat shock proteins released by ischemic cells and during tissue damage, like myocardial infarction or collateral remodeling, and initiate a proinflammatory response thus playing a decisive role in angiogenesis and arteriogenesis (Termeer and others 2002; Arslan and others 2011).
Mononuclear Cells in Angiogenesis and Arteriogenesis
Neovascularization is the general term or the development of new blood vessels from the existing vasculature by angiogenesis or arteriogenesis induced by therapeutical, physiological, or pathological stimulation. In contrast, vasculogenesis is defined as the development of de novo blood vessels from endothelial stem cells in the absence of existing vessels (Simons 2005; Faber and others 2014). Here, we focus on the 2 processes of blood vessel development that can be clearly distinguished after birth, namely angiogenesis and arteriogenesis. Angiogenesis is the sprouting of new capillaries from preexisting postcapillary venules in an ischemic area, whereas arteriogenesis describes the development of mature arteries from preexisting nonfunctional arterioles after increased blood flow due to proximal arterial lumen narrowing or arterial obstruction (Conway and others 2001; van Royen and others 2001; de Groot and others 2009; Schirmer and others 2009; van der Laan and others 2009; Faber and others 2014). Arteriogenesis is reparative by nature, restoring hampered blood flow in the setting of arterial obstructive disease. During coronary artery disease (CAD) obstruction of the main coronary arteries leads to a difference in the pressure distal and proximal to the obstruction. As blood finds its way from the highest pressure to the lowest pressure, it will circumvent the obstructed artery, thereby creating a natural bypass. During the early phase of arteriogenesis, the blood flows from the blocked artery into preexisting arterioles (Heil and Schaper 2007; de Groot and others 2009). The increased blood flow over the preexisting arterioles increases the shear stress, which is the initial trigger of arteriogenesis.
This shear stress directly activates the endothelium by increasing the expression levels of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) (Hoefer and others 2004; Luo and others 2011), and platelet endothelial cell adhesion molecule-1 (PECAM-1) (Chen and others 2010). Additionally, monocyte chemoattractant protein-1 (MCP-1) is induced, a potent chemoattractant for monocytes that interact with the chemokine (C-C motif) receptor 2 (CCR2) on monocytes (Hoefer and others 2005; Shireman 2007). Monocyte recruitment plays a major role in initiating arteriogenesis (Hoefer and others 2005; Shireman 2007). Monocytes are recruited to the intima of the vessel, where they differentiate into macrophages that produce growth factors and cytokines, including transforming growth factor-β1 (TGF-β1), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), and MCP-1. These factors are known to enhance vessel remodeling by inducing the proliferation of vascular smooth muscle cells (SMCs) and ECs, and by attracting more monocytes (Shireman 2007; Schirmer and others 2009). Once inside the intima, macrophages degrade the internal elastic lamina by the secretion of matrix metalloproteinase (MMPs), enabling cell movement and general tissue reshaping for arterial growth (Galis and others 2002). Finally, a prolonged state of increased shear stress along the preexisting arteriole results in a mature conduction vessel that is able to circumvent the obstructed artery (Schaper and Scholz 2003; de Groot and others 2009).
In addition to monocytes, several other bone marrow-derived cells have been implicated in the initiation and progression of arteriogenesis. A role for endothelial progenitor cells (EPCs) in arteriogenesis has been described by Urbich and others (2003). Together with more recent evidence, these studies indicate that EPCs are a subset of proangiogenic monocytes of distinct myeloid origin (Medina and others 2011; Urbich and others 2011; van der Pouw Kraan and others 2012). Experimental models of hind limb ischemia have shown additional potentiating effects of CD4+ and CD8+ T cells and NK cells in the initiating phase of arteriogenesis (Stabile and others 2003, 2006; van Weel and others 2007).
Effects of Immune Modulation by IFN-β on Angiogenesis
Angiogenesis refers to the sprouting of a new vasculature from preexisting vessels (Conway and others 2001; van der Laan and others 2009). Angiogenesis can be physiologic during female reproduction and wound healing, but it can also be detrimental in pathologies like atherosclerosis and tumor growth. Angiogenesis is required for tumor growth by delivering oxygen and nutrients to the rapidly proliferating tumor cells (Fidler and others 2002). McCarty and others (2002) showed that endogenous IFN-α/IFN-β is involved in the regulation of angiogenesis. Gelfoam sponges were filled with the proangiogenic molecules bFGF, VEGF, and TGF-α and implanted in IFN-α/-β receptor-deficient mice, resulting in a significantly increased vasculature when compared to identical sponges implanted in control mice. This indicates that IFN-α/-β signaling is involved in the inhibition of angiogenesis (McCarty and others 2002). Collectively, the antiangiogenic effects of type I IFNs resulted in tumor growth regression in experimental animal models (McCarty and others 2002; Borden and others 2007; Indraccolo 2010).
Several mechanisms have been proposed by which IFN inhibits angiogenesis. Jablonska and others (2010) showed that endogenous IFN-β may inhibit tumor growth and angiogenesis through its effect on tumor-infiltrating neutrophils. They showed that IFN-β-deficient mice injected with tumor cells have faster growing tumors and a larger tumor size when compared to wild-type (WT) mice. This is accompanied by an increased tumor angiogenesis and elevated levels of tumor-infiltrated CD11b+Gr1+ neutrophils, which express high levels of proangiogenic and homing factors. IFN-β treatment of these tumor-infiltrating neutrophils leads to reduced gene expression levels of VEGF, MMP9, and C-X-C chemokine receptor type 4 (CXCR4), the receptor for stromal-derived factor-1 (SDF-1). Depletion of IFN-β-responsive CD11b+Gr1+ neutrophils reduced tumor growth and angiogenesis in both control and IFN-β-deficient mice. Supporting this fact, normal mice injected with tumor cells and neutrophils obtained from tumor-bearing IFNAR-deficient mice showed increased tumor growth and developed more mature vessels when compared with neutrophils obtained from tumor-bearing WT mice. In conclusion, in this study neutrophils represent the main cell population for tumor angiogenic activity and are controlled by endogenous IFN-β (Jablonska and others 2010). Piccard and others reviewed the differences in the N1 and N2 neutrophil phenotypes. N1 neutrophils have an antitumoral function whereas the N2 neutrophils have a protumoral function. IFN-β stimulates the antiangiogenic N1 phenotype and inhibits the angiogenic N2 phenotype (Piccard and others 2012).
IFN-β may also reduce the number of nonfunctional vessels in tumors. Dickson and others (2007) showed that continuous systemic delivery of human IFN-β, transgenically expressed by an AAV-vector, resulted in a sustained maturation of the intra-tumor vasculature of human xenografts in immunodeficient mice while angiogenesis was inhibited. Because human IFN-β has little effect on the murine IFN receptor, this stabilizing effect could not have been mediated by the mouse vasculature itself. Instead, IFN-β-mediated release of angiopoetin-1 (ANG-1) by the human xenografts was responsible for the recruitment of perivascular cells to the tumor vessels leading to stabilized vessels with a significantly reduced permeability. The role of Ang-1 in vessel stabilization was also reported elsewhere (Stoeltzing and others 2003). In a study by Cao and others (2001) tumor cells were transduced with an adenoviral vector encoding the murine IFN-β gene (AdIFN-β) injected in nude mice to suppress tumorigenicity. AdIFN-β inhibited tumor growth and angiogenesis in a dose-dependent manner. At a high dose of AdIFN-β, these tumors showed elevated expression of inducible nitric oxide synthase, combined with reduced levels of bFGF and TGF-β1 (Cao and others 2001). According to Cao and others IFN-β inhibits tumor growth directly by inducing apoptosis, and indirectly by NO-mediated suppression of bFGF and TGF-β1 expression in tumor cells. In addition, NO dose-dependently blocks bFGF-induced proliferation of ECs in vitro, which may have contributed to the inhibition of angiogenesis as well (RayChaudhury and others 1996; Cao and others 2001). Altogether, these studies indicate that IFN-β indirectly inhibits tumor angiogenesis through a cascade that upregulates NO, leading to a reduced bFGF expression and responsiveness followed by a reduced bFGF-induced proliferation of ECs.
Furthermore, endogenous NO decreases the adhesiveness of ECs for monocytes and reduces the expression and secretion of MCP-1 by ECs, leading to inhibition of monocyte migration (Zeiher and others 1995; Boger and others 2000).
A more recent study showed that EPCs, currently known as circulating angiogenic cells of monocytic origin, can enhance tumor angiogenesis resulting in tumor growth. Intriguingly, transfection of EPCs with the IFN-β gene counteracted the tumor-progressive function of EPCs leading to reduced tumor growth and angiogenesis due to a diminished secretion of VEGF (Xiao and others 2012). In addition, IFN-β inhibits angiogenesis in experimental glioma, which may be the direct consequence of the inhibition of VEGF secretion by glioma cells and the inhibition of glioma cell-induced endothelial cell (EC) migration (Takano and others 2014).
IFN-β may also exert antitumor effects through its effects on T cells. Oh and others (2012) showed that the reduced tumor growth in vivo as a result of injecting IFN-β-producing adenovirus was dependent on CD8+ T cells. IFN-β-treated mice showed enhanced proliferation of antigen-specific CD8+ T cells, which are held responsible for tumoricidal activities. The role of IFN-β is described in more detail for CD4+ T helper cells, especially in relation to the subset of T helper 17 cells (Th17). Th17 cells produce the proinflammatory cytokine IL-17 that possesses potent proangiogenic activity, playing a pivotal role in tumor activities (Liu and others 2011) and during angiogenesis in the murine hind limb arterial ligation model (Hata and others 2011). Th17 cells in combination with IFN-β were used in an in vivo study (Sweeney and others 2011) that showed that IFN-β treatment inhibited Th17 cell differentiation through induction of IL-27. Interestingly, a study by Shimizu and others (2006) demonstrated that IL-27 has potent antiangiogenic and antitumor capacities. Therefore, IFN-β-induced IL-27 may be responsible for the inhibition of IL-17 production, resulting in impaired angiogenesis. In line with the multiple mechanisms by which IFN-β regulates angiogenesis, Taylor and others (2008) suggested that not a single gene, but most likely multiple IFN-β response genes are responsible for the inhibition of angiogenesis.
At present it is unknown, how type I IFN affects angiogenesis within the atherosclerotic plaque (a feature associated with plaque instability). It can be anticipated that type I IFN may inhibit angiogenesis within plaques, analogous to other anatomical locations. However, type I IFN may also lead to the development of vulnerable plaques by inducing increased macrophage accumulation and development of larger necrotic cores (independent of lesion size) (Goossens and others 2010). Absence of endogenous type I IFN signaling in myeloid cells inhibits lesion development, protects against lesional accumulation of macrophages, and prevents necrotic core formation and thus directly affects plaque stability.
The atherosclerosis-promoting effect of type I IFN was confirmed in a study in which IFN-α treatment resulted in increased lesion size and macrophage accumulation (Doring and others 2012). Furthermore, activation of plasmacytoid dendritic cells (pDCs, specialized cells to produce IFN-α) increased atherosclerotic lesion development and macrophage accumulation, which could be prevented by depletion of pDCs. The overlapping effects of IFN-α and IFN-β on atherosclerosis are not surprising as they both signal through the same receptors.
Molecular Effectors of IFN-β in Angiogenesis and Arteriogenesis
In addition to its well-documented role in angiogenesis, a decisive role for IFN-β in arteriogenesis has recently emerged. TLR4-stimulated monocytes from patients with poorly developed collateral arteries display increased type I IFN reactivity. These findings were corroborated in the murine hind limb arterial ligation model for arteriogenesis, where exogenous application of IFN-β was associated with reduced collateral artery formation (Schirmer and others 2008), while enhanced collateral artery formation is observed in IFNAR-1−/− mice (Schirmer and others 2010). Significantly elevated expression levels of IFN-β and IFN-β-downstream genes were found in monocytes from patients with a partial occlusion of coronary arteries, including IL-27, chemokine (C-X-C motif) ligand (CXCL)-10, and CXCL11. This was corroborated in LPS-stimulated monocytes from an independent cohort of patients with chronic total coronary occlusion and a poor collateral circulation (van der Laan and others 2012). The chemokine CXCL subfamily is able to induce chemotaxis and tissue extravasation, and could modulate leukocyte behavior in the context of inflammation (Groom and Luster 2011). Based on these findings, we will now discuss the role of IFN-β and its downstream genes in angiogenesis and translate these findings to arteriogenesis.
IFN-β directly affects vascular cell proliferation and survival
It has been described that IFNs can lead to apoptosis of cells after viral infection (Stetson and Medzhitov 2006; Vilcek 2006; Borden and others 2007; Zhang and others 2008b). Additionally, IFN-β is therapeutically used to eliminate cancer by its antitumor and antiangiogenic activity, as described above. Because EC integrity and proliferation is important during both angiogenesis and arteriogenesis, the effects of IFN-β on EC may be equally relevant for both processes. IFN-β may affect EC survival (Izawa and others 2002). Inhibition of tumorigenicity after human adenoviral IFN-β gene therapy resulted in an indirect apoptotic effect of IFN-β on ECs through downregulation of as yet unspecified “endothelial cell survival factors.” Additionally, type I IFN directly affects EC function as indicated by the IFN-induced inhibition of migration, chemotaxis, and proliferation of human umbilical vein endothelial cells (HUVEC) (Yoshida and others 1996; Park and others 2006). Erdmann and others (2011) described that IFN-β induced a cell cycle arrest and inhibited proliferation of micro-human lung microvascular endothelial cells and macrovascular (HUVEC) ECs but apoptosis was, surprisingly, inhibited. The authors therefore concluded that IFN-β has no direct cytotoxic effect on ECs. Furthermore, Schirmer and others (2010) showed that activation of IFN-β signaling leads to reduced vascular SMC proliferation (Fig. 1). Additionally, Zhang and others (2008a) demonstrated in apolipoprotein E-deficient mice that IFN-β is able to prevent angiotensin II-induced vascular SMC proliferation after ligation of the carotid artery. In a study by Stephan and others (1997) it was reported that gene transfer of IFN-β also inhibited vascular SMC proliferation and intimal hyperplasia in a porcine balloon injury artery model. Thus, a general growth inhibitory effect of IFN on vascular cell proliferation is apparent from a variety of studies.
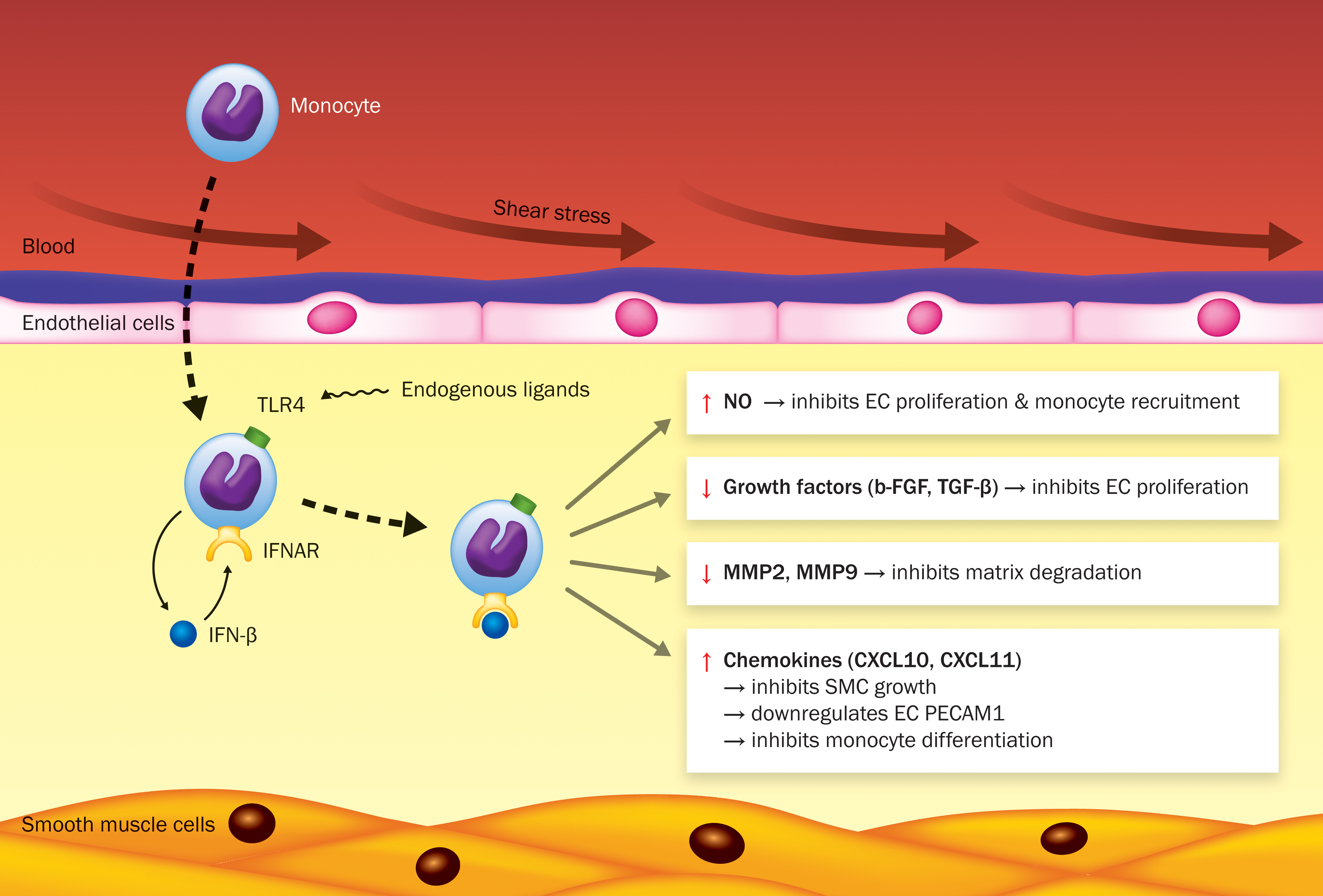
Mechanisms of the inhibiting effect of interferon (IFN)-β on arteriogenesis. Shear stress activates the endothelium, thereby increasing the expression of adhesion molecules, which is important for monocyte recruitment into the intima during arteriogenesis. Recruited monocytes differentiate into macrophages, which produce growth factors, chemokines, and proteases. These factors promote vessel remodeling by inducing vascular smooth muscle cell (SMC) and endothelial cell (EC) proliferation, attraction of more monocytes, and degradation of extracellular matrix (ECM) proteins. Danger-associated molecular patterns (DAMPs), activating Toll-like receptor (TLR4) present on monocytes/macrophages can induce IFN-beta (IFN-β) production by recognizing endogenous ligands, such as degradation products of ECM. Released IFN-β may interact in both a paracrine and/or autocrine manner with the IFN-α/β receptor (IFNAR) leading to the activation of the IFN-β signaling pathway. IFN-β has pleiotropic functions by displaying antiproliferative and immunomodulatory properties during arteriogenesis and angiogenesis. Activation of IFN-β signaling induces nitric oxide (NO) production, leading to reduced growth factor expression [basic fibroblast growth factor (bFGF) and transforming growth factor-β1 (TGF-β1)] and subsequently reduced proliferation of ECs. NO also suppresses monocyte recruitment. IFN-β signaling is also involved in reduced matrix metalloproteinases (MMP) 2 and 9 expression, suppressing matrix degradation. Furthermore, IFN-β signaling may lead to reduced vascular SMC proliferation. IFN-β-induced chemokine (C-X-C motif) ligand (CXCL)-10 may downregulate endothelial platelet EC adhesion molecule-1 (PECAM-1) expression, which may result in impaired shear stress sensing and monocyte transmigration. Another IFN-β-induced chemokine, CXCL11 may impair the differentiation of monocytes. Color images available online at
IFN-β inhibits expression of growth factors
Cao and others (2001) demonstrated that reduction of tumor angiogenesis by IFN-β was triggered by an increased production of NO, which was accompanied by reduced growth factor levels such as bFGF and TGF-β1. Both bFGF and TGF-β1 have been shown to induce collateral vessel growth (van Royen and others 2002; Wu and others 2010). Since IFN-β is involved in the reduction of bFGF and TGF-β1 during tumor angiogenesis, it might also impair arteriogenesis by an NO-mediated reduction of bFGF and TGF-β1 levels (Fig. 1). Additionally, NO also inhibits bFGF-induced EC proliferation and monocyte migration (Zeiher and others 1995; RayChaudhury and others 1996; Boger and others 2000).
According to Zheng and others (2011), the proangiogenic endothelial growth factor VEGF is responsible for the degradation of IFNAR-1, thus resulting in the promotion of angiogenesis. This degradation is initiated by the phosphorylation of IFNAR-1 followed by ubiquitination, which is induced by the activation of protein kinase D2 (PKD2). Current clinical trials with ectopic growth factor delivery have shown disappointing results, as reviewed by Schirmer and others (2009). Given the evidence from experimental models, this may result from an inhibitory interplay with endogenous IFN-β affecting cell survival and growth.
IFN-β affects cytokine and chemokine levels
Exaggerated induction of IL-27, CXCL10, and CXCL11 gene expression was found in LPS-stimulated monocytes from patients with poor collateral vessel formation (Schirmer and others 2008). The role of IL-27 during collateral formation is largely unknown, but its inhibitory activity on both proangiogenic Th17 cells and angiogenesis itself may indicate a similar regulating role in arteriogenesis. CXCL10 and CXCL11, and CXCL9, are regarded angiostatic chemokines and share a common receptor CXCR3 (Romagnani and others 2004). A role of CXCL10 in downregulating PECAM-1 expression has been studied by Glaser and others (2004), showing that CXCL10 neutralization resulted in elevated PECAM-1 expression levels in a spinal cord injury mouse model. PECAM-1 is one of the major mechanosensors for shear stress (Tzima and others 2005). Moreover, PECAM-1 plays a role in the interaction between blood cells (eg, platelets, monocytes, and neutrophils) and ECs, preceding their transmigration into the intima (Chen and others 2010). As a result PECAM-1 plays an essential role in arteriogenesis. Thus, downregulation of PECAM-1 on endothelium by CXCL10 can result in (1) lack of shear stress sensing and (2) impaired monocyte transmigration to the growing collaterals (Fig. 1). In contrast, CXCL10 may not affect arteriogenesis at other stages, since CXCL10 stimulation of vascular SMCs in vitro did not affect their proliferation (Schirmer and others 2010). Surprisingly, CXCL10 deficiency impairs arteriogenesis in the murine hind limb arterial ligation model, without affecting inflammation. In this study a reduced migration of vascular SMCs was held responsible for the impaired arteriogenic response (van den Borne and others 2014a). Extrapolation of these experiments to the human situation is hampered by the species differences in CXCL10 receptor usage. Human CXCR3 has 3 splice variants (CXCR3-A, -B, and -alt), while mice only express the CXCR3-A isoform (Billottet and others 2013). The human splice variant CXCR3-B is expressed on ECs during the S/G2-M phase of the cell cycle and exerts angiostatic effects (inhibition of proliferation, enhancement of apoptosis, and reduced tube-like vessel formation), while the CXCR3-A isoform exerts proangiogenic effects, and it is the only isoform in mice (Romagnani and others 2001, 2004; Billottet and others 2013; van den Borne and others 2014b). Therefore, CXCL10 deficiency in mice affects only the proangiogenic effect through CXCR3-A, while in humans CXCL10 exerts both pro- and antiangiogenic effects.
CXCL11 may influence arteriogenesis by a mechanism similar to CXCL10, as both chemokines bind the same receptors, CXCR3A and CXCR3B (Billottet and others 2013). Interestingly, in atherosclerosis-prone mice (ApoE−/−), an improved hind limb collateral formation is associated with reduced local expression levels of CXCL10 and CXCL11 (Schirmer and others 2012), suggesting an inhibitory role in collateral remodeling in hypercholesterolemic mice. Coelho and others (2005) described that IFN-β-induced gene expression of CXCL11 and recombinant CXCL11 itself, affected the differentiation of primary human monocytes during osteoclastogenesis. This could imply that CXCL11 also impairs the differentiation of monocytes during collateral vessel formation (Fig. 1). In a study performed by Kao and others (2003) it was found that patients who underwent heart transplantation and developed severe transplant CAD (TCAD), had higher CXCL11 levels in both serum and in ECs from TCAD lesions. Additionally, immunohistochemical stainings showed infiltration of CXCR3-positive mononuclear cells within the TCAD lesions, which can clarify the pathogenesis of the disease. It was suggested that CXCL11 might act as a biomarker to diagnose patients with a high risk for TCAD. Therefore, CXCL11 is a potential suitable target for intervention, as its inhibition may increase arteriogenesis and decrease atherosclerosis.
IFN-β affects tissue-degrading enzymes
According to Cai and others (2003) the development of arteriogenesis can be subdivided into 3 phases, the early phase, (active) growth phase and the final maturation phase. Proteins involved in all 3 phases include the highly expressed adventitial MMP2 and MMP9, which degrade extracellular matrix proteins collagen and elastin thus initiating the enlargement of collateral vessels. Ma and others (2001) revealed that IFN-γ and IFN-β both inhibited MMP9 gene transcription, resulting in reduced MMP9 expression levels in vitro in a STAT-1-dependent manner. This finding is in accordance with the study performed by Nelissen and others (2002) in which it was shown that IFN-β inhibited the production of MMP9 in monocytic cell lines and peripheral blood mononuclear cells. Moreover, it was shown that MMP2 and MMP9 expression levels were reduced in STAT-1-reconstituted tumors (Huang and others 2002). Since IFN-β is involved in the reduction of MMP expression, it might impair tissue degradation during arteriogenesis (Fig. 1).
Conclusion
IFN-β plays a pivotal inhibitory role in angiogenesis and arteriogenesis. IFN-β inhibits angiogenesis (1) by its effect on tumor-infiltrating neutrophils by reducing gene expression of VEGF, MMP9, and CXCR4, and inhibition of the angiogenic neutrophil N2 phenotype, (2) by a direct effect on ECs, by inducting apoptosis, and preventing migration, chemotaxis, and proliferation, (3) by an indirect mechanism mediated by NO-induced suppression of bFGF and TGF-β1.
The inhibitory effect of IFN-β on arteriogenesis may occur by multiple IFN-β-mediated mechanisms as summarized in Fig. 1. The accumulating evidence on the regulatory role of many of the IFN-induced genes in angiogenesis, their emerging roles in arteriogenesis, and a recent report that IFN-β actually accelerates atherosclerosis (Goossens and others 2010) makes IFN-β inhibition a challenging novel approach to relief ischemic heart disease.
Footnotes
Acknowledgments
This work was supported by an Institutional grant from ICaR-VU of the VU University Medical Center, Amsterdam, The Netherlands (A.J.G.H., T.P.K.). The authors would like to thank Ico Davids for designing the figure in this review.
Author Disclosure Statement
No competing financial interests exist.