
Abstract
Although endothelial cell (EC) infection is not widespread during dengue virus (DENV) infection in vivo, the endothelium is the site of the pathogenic effects seen in severe DENV disease. In this study, we investigated DENV infection of primary EC and defined factors that influence infection in this cell type. Consistent with in vivo findings where EC infection is infrequent, only 3%–15% of EC became productively DENV-2-infected in vitro. This low level infection could not be attributed to inhibition by heparin, EC donor variation, heterogeneity, or biological source. DENV-infection of EC was associated with induction of innate immune responses, including increased STAT1 protein, STAT1- phosphorylation, interferon (IFN)-β, OAS-1, IFIT-1/ISG56, and viperin mRNA. Antibody blocking of IFN-β inhibited the induction of OAS1, IFIT1/ISG56, and viperin while shRNA knockdown of viperin enhanced DENV-infection in EC. DENV-infection of EC resulted in increased activity of sphingosine kinase 1, a factor important in maintaining vascular integrity, and altered basal and stimulated changes in barrier integrity of DENV-infected EC monolayers. Thus, DENV productively infects only a small percentage of primary EC but this has a major influence on induction of IFN-β driven innate immune responses that can restrict infection while the EC themselves are functionally altered. These changes may have important consequences for the endothelium and are reflective of pathogenic changes associated with vascular leakage, as seen in DENV disease.
Introduction
D
Many studies have described low level DENV-infection of primary EC (Anderson and others 1997; Bunyaratvej and others 1997; Diamond and others 2000a; Huang and others 2000; Warke and others 2003; Arevalo and others 2009; Liu and others 2009) or EC lines in vitro (Peyrefitte and others 2006). More recently, up to 90% of human umbilical vein endothelial cells (HUVEC) were DENV infected when heparin, a common inclusion in culture medium following isolation of primary HUVEC, was removed from culture (Dalrymple and Mackow 2011), suggesting most HUVEC are susceptible to DENV but infection is inhibited by heparin. Microarray analysis has demonstrated upregulation of a number of cytokines, chemokines, and interferon-stimulated genes (ISGs) in DENV-infected EC (Warke and others 2003; Dalrymple and Mackow 2012a). Antibody blocking studies have suggested that it is the action of interferon (IFN)-β that induces these ISGs and limits DENV infection (Dalrymple and Mackow 2012a). Thus, the literature is conflicting on the degree of susceptibility of ECs to DENV infection, but suggests that EC can be infected by DENV that induces an inflammatory response in these cells with a central role for IFN-β.
In this study we have extended this understanding of DENV infection of EC. Using primary ECs derived from 2 different sources and multiple EC donors we demonstrate a productive DENV-2 infection in only a small percentage of cells. DENV-infection of EC was accompanied by early innate immune responses, including the induction of IFN-β mRNA and the ISG viperin, which acts to restrict viral infection. Importantly, even in the face of only a small percentage of productively DENV-infected EC, we observe a clear change in cellular protein and mRNA profiles related to the host innate immune response and altered EC function. Our data suggest that EC may not be a major target for DENV infection but the responses to this low level DENV infection in EC are reflective of pathogenic changes in the vasculature and may have important consequences for vascular leakage and DENV disease in vivo.
Materials and Methods
HUVEC and endothelial colony forming cells
Human umbilical cords and cord blood were collected with informed consent and in line with Central Northern Adelaide Health Service human ethics approval and in accordance with the “Declaration of Helsinki”. Primary HUVEC were isolated by collagenase digestion of human umbilical veins, cultured on gelatin (Sigma)-coated flasks in M199 media (Sigma) supplemented with 20% (v/v) fetal calf serum (FCS; Hyclone), penicillin, streptomycin, 20 mM HEPES, 1.5% (w/v) sodium pyruvate (all Gibco), nonessential amino acids (Sigma), and EC growth supplement (BD Bioscience) (HUVEC medium) as previously described (Litwin and others 1997). HUVEC were utilized in infection studies at passage 1–4.
Endothelial colony forming cells (ECFC), formerly known as late outgrowth endothelial progenitor cells were isolated by CD133 magnetic bead isolation of cells from human umbilical cord blood collected and processed as previously described (Appleby and others 2012). CD133− mononuclear cells were then cultured in EGM-2 endothelial growth media (Lonza) supplemented with an EGM2 bullet kit (Lonza) without heparin and 20% (v/v) FCS. Further culture, isolation of colonies and passaging occurred as previously described (Ingram and others 2004). ECFC were utilized at passages 4–8.
DENV infection and plaque assay
Cells were infected at a multiplicity of infection (MOI) of 1 for 90 min using Mon601, a laboratory clone of the DENV type-2 (DENV-2) strain New Guinea C (Gualano and others 1998). Virus stocks were produced from in vitro transcribed RNA that was transfected into baby hamster kidney (BHK-21) cells and supernatant amplified in C6/36 insect cells. Titer of virus stocks and cell culture supernatant samples was determined by plaque assay on Vero, African Green monkey kidney cells. Plaques were detected by neutral red overlay and quantitated as plaque forming units (pfu) per mL. For IFN-β blocking studies HUVEC were infected at an MOI of 1 and then cultured postinfection (pi) in media supplemented with control or neutralizing IFN-β antibody (Pbl interferon source) at 1,000 U/mL.
RNA extraction and reverse transcription-polymerase chain reaction
Total RNA was extracted from cells using TRIzol (Gibco) and subjected to reverse transcription using random hexamers (NEB). cDNA was subjected to real time PCR as follows: DENV primers, 5.1 GCAGATCTCTGATGAATAACCAAC and 3.2: TTGTCAGCTGTTGTACAGTCG; IFN-β primers, TGTCAACATGACCAACAAGTGTCT and GCAAGTTGTAGCTCATGGAAAGAG; OAS-1 primers, TCCACCTGCTTCACAGAACTACA and GGCGGATGAGGCTCTTGAG; IFIT-1 primers, AACTTAATGCAGGAAGAACATGACAA and CTGCCAGTCTGCCCATGTG; viperin primers, GTGAGCAATGGAAGCCTGATC and GCTGTCACAGGAGATAGCGAGAA. Semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) was performed by amplification of a standard curve in parallel and results were normalized against cyclophilin quantitated using primers GGCAAATGCTGGACCCAACACAAA and CTAGGCATGGGAGGGAACAAGGAA. Reactions were performed in a Corbett Rotorgene 6000 using sybergreen reaction mix (iTaq; BioRad).
Immunostaining and confocal microscopy
Cells were grown on gelatin-coated coverslips, fixed in 4% (w/v) paraformaldehyde and stored at 4°C. Fixed cells were permeabilized, blocked, and immunostained for dsRNA (goat anti-mouse antibody at 1/200 dilution; English and Scientific Consulting, J2), DENV antigen (DENV patient sera at 1/5,000 dilution), as described previously (Carr and others 2013). Bound antibody was detected with goat-anti mouse-555 (red) or goat-anti human-488 (green), nuclei were stained with Hoechst (5 μg/mL, blue) and images captured by confocal microscopy (Leica Sp5 Spectral Confocal Microscope).
Crystal violet staining
Cell were fixed and stained in 50% methanol containing 0.2% (w/v) crystal violet. Unbound stain was removed and fixed cells washed extensively with water. Bound stain was solubilized with 2% (w/v) sodium dodecyl sulfate (SDS) and absorbance at 540 nm was quantitated (Titertek Twinreader).
SDS-polyacrylamide gel electrophoresis and western blotting
Cells were lysed in passive lysis buffer (Promega) with protease inhibitors (Roche CompleteMini) and protein levels quantitated (Biorad). Fifteen microgram of total protein was subjected to SDS-polyacrylamide gel electrophoresis, proteins transferred to nitrocellulose membranes and membranes serially probed for STAT1-Y701 (Cell Signalling), STAT1-S727 (Cell Signalling), total STAT1 (Cell Signalling), and actin (Millipore). Bound antibody complexes were detected by chemiluminesence (Biorad), images captured (LAS4000; Fuji Film) and quantitated using Image J (Schneider and others 2012).
shRNA production and infection of HUVEC
Viperin and control shRNA lentivirus was made as previously described (Helbig and others 2011). Following filtration of cultured media from lentivirus-transduced cells through a 0.45 μm filter, the viral supernatants were diluted 1:3 in HUVEC media (M199 media containing the supplements described above) and incubated with HUVEC for 8 h. Lentivirus containing supernatant was removed and replaced with fresh HUVEC media and cells incubated for approximately a further 64 h, prior to DENV infection, as above.
Sphingosine kinase 1 activity assay
Cells were lysed and sphingosine kinase 1 (SK1) activity quantitated by transfer of 32P to sphingosine in vitro, as described previously (Pitman and others 2012; Carr and others 2013). Total protein was quantitated (Biorad protein assay) and results expressed as SK1 activity/μg of protein.
EC permeability assay
Cells were DENV or mock infected (heat inactivated virus, 80°C, 20 min) at an MOI=1, as described above, trypsinized, and replated in complete HUVEC medium at 15,000 cells/well in 16 well E-view plates (ACEA Biosciences, Inc.). Cells were allowed to recover in complete HUVEC media and media changed at 2–3 h post plating. Plates were incubated and the cell index measured by an impedence-based real-time cell analysis using the x-celligence system (ACEA Biosciences, Inc.). Prior to plating, E-plates were normalized with medium alone and measurements were taken every hour. In relevant experiments, at 3 h pi, media was changed to M199+0.1% (v/v) FCS, without growth factors and cells incubated overnight. At 24 h pi media was changed to test stimulus (i) complete HUVEC media or (ii) M199+0.1% (v/v) FCS+0.5 ng/mL recombinant human tumor necrosis factor (TNF)-α (ProSpec). Cells were incubated for a further 12 h and cell index measured hourly.
Statistical analysis
Normally distributed data sets were compared using an unpaired Student's t-test. Experiments were replicated as indicated in each figure legend. Statistical analysis was performed using Microsoft Excel.
Results
DENV productively infects only a small percentage of EC including ECFC
Recent studies have shown that heparin inhibits DENV infection of HUVEC and that by 24 h pi, the majority of HUVEC become DENV-antigen-positive (Dalrymple and Mackow 2011). To assess this in freshly isolated primary HUVEC, we cultured these cells in the presence or absence of heparin, DENV-infected and measured infectious virus release. Results confirm increased DENV-infection in the absence of heparin (Fig. 1A), but still only a low percentage of the total cell population was DENV-antigen positive (Table 1). DENV-infection at a higher MOI of 5 produced a modest increase in infectious virus release (data not shown) and HUVEC showed slightly higher infected cell numbers when using an alternative DENV-2 strain, POU-312 (Table 1). Overall, however, the percentage of DENV-antigen-positive cells was still low and in the order of 3%–15% (Table 1).
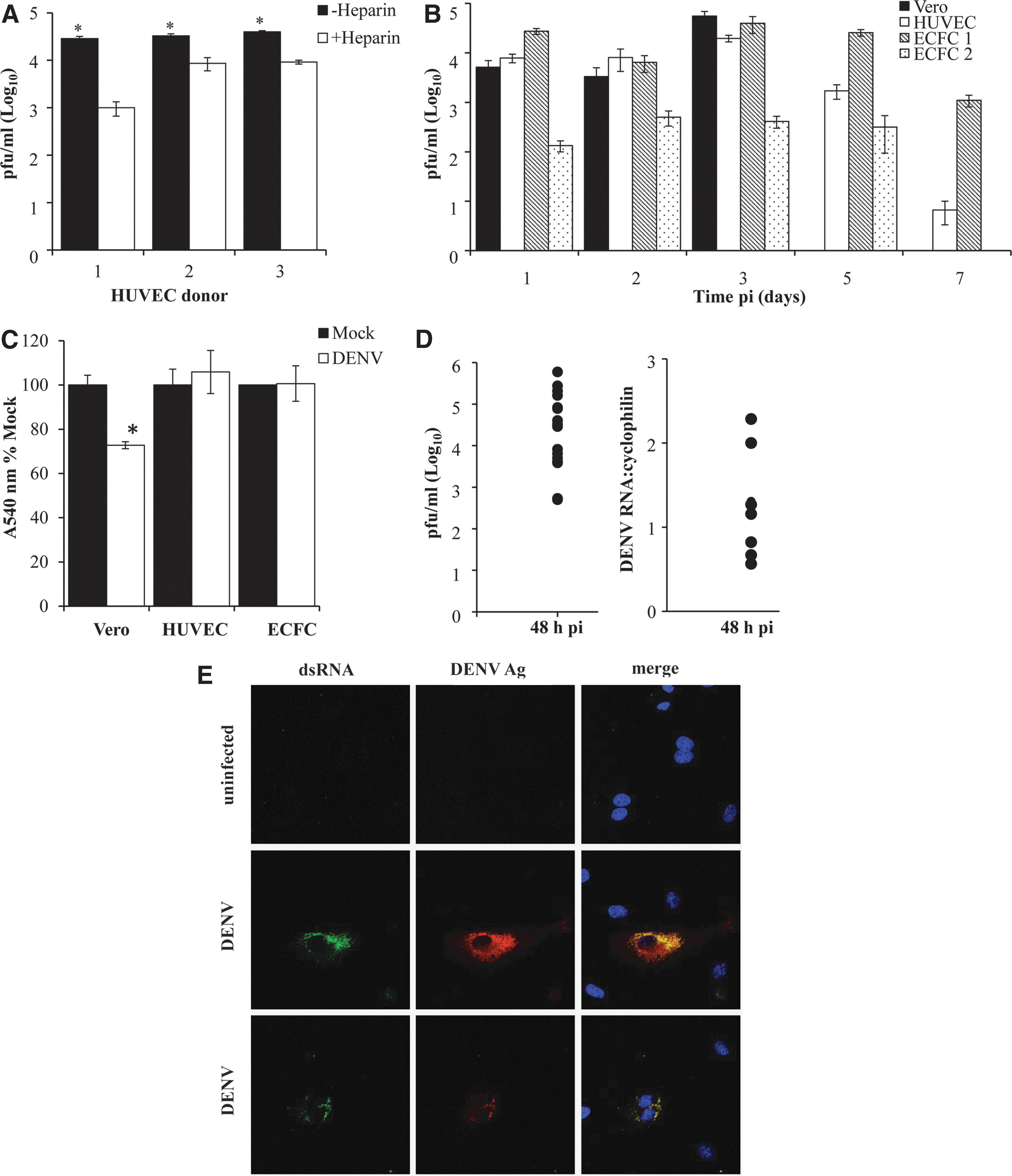
Dengue virus (DENV)-2 productively infects endothelial cell (EC). Human umbilical vein endothelial cells (HUVEC) or endothelial colony forming cells (ECFC) were isolated and mock or DENV-2 infected at a multiplicity of infection (MOI)=1.
Cell counting was performed on n=2–10 fields with>150 cells.
DENV, dengue virus; HUVEC, human umbilical vein endothelial cells; ECFC, endothelial colony forming cells.
This low susceptibility of EC to DENV-2 infection noted above could be due to donor variation, heterogeneity in the HUVEC population, or variation in EC susceptibility due to the location in the body they are isolated from. To address this we next DENV-infected primary HUVEC, ECFC and Vero cells, a control DENV-susceptible cell type and quantitated DENV-infection. ECFC represent a second novel source of primary EC and are clonally derived from a single circulating EC progenitor. Both HUVEC and ECFC demonstrated productive DENV-infection, comparable to Vero cells (Fig. 1B). DENV-infection was highest early (days 24–72 h, pi) and persisted but declined with time. No cytopathic effect (CPE) was associated with DENV-infection of primary HUVEC or ECFC cultures, visually or as quantitated by crystal violet vital dye staining (Fig. 1C). In contrast, DENV-infection of Vero cells produced infectious DENV (Fig. 1B) but by day 3 pi, a clear visual DENV-induced CPE was observed with a reduction in vital dye staining (Fig. 1C).
The level of DENV-2 infectious virus release was comparable in cell preparations from different EC donors, although some donor variation was observed (Fig. 1B, D). DENV-infected cultures from multiple donors supported active DENV-2 replication, as measured by high DENV titers and the presence of DENV-2 RNA at day 2 pi (Fig. 1D). Additionally, dual DENV-2 antigen and dsRNA positive EC were observed by immunostaining and fluorescence microscopy (Fig. 1E). The DENV-infection was restricted to a small number of cells and did not spread throughout the entire culture (Fig. 1E). The number of DENV-infected cells was quantitated from multiple EC donors and observed to be in the range of 3%–15% (Table 1), much lower than the infection of the majority of Vero cells under the same conditions (data not shown). Thus, primary EC from 2 different sources and multiple donors are susceptible to a low level productive DENV-2 infection in vitro.
DENV infection of EC induces innate immune responses that restrict DENV-2 infection
To assess the ability of DENV-2 to initiate an innate immune response in EC we investigated the phosphorylation status of STAT1, a key molecule in IFN signaling. Results demonstrate a clear time and MOI-dependent induction of STAT1-Y701 and S727 phosphorylation in DENV-infected EC from 24 h pi and still elevated at 72 h pi (Fig. 2). Interestingly, we also observed an increase in total STAT1 protein (Fig. 2). We next assessed the upregulation of IFN-β mRNA and a number of known antiviral effector ISGs (IFIT-1/ISG56, OAS-1, and viperin) in response to DENV-2 infection of EC. DENV infection was confirmed by the detection of DENV RNA, which increased from 24 to 72 h pi (Fig. 3A) and was detected in all cases and in multiple EC donors at 48 h pi (Fig. 3B). The levels of IFN-β, IFIT-1/ISG56, OAS-1, and viperin mRNAs were undetectable or negligible in mock-infected cells (Fig. 3). IFN-β was induced in DENV-infected cells at 24 h pi and still present at 72 h pi (Fig. 3A). Levels of IFN-β mRNA, were elevated at 48 h pi in all cases and in multiple EC donors (Fig. 3B). In contrast, mRNA for IFIT1/ISG56, OAS-1, and viperin were all induced in DENV-infected cells at 24 h pi, further elevated at 48 h pi but declining by 72 h pi (Fig. 3A). Again elevated mRNA levels at 48 h pi were observed in all cases and in multiple EC donors (Fig. 3B). These results indicate that a robust innate antiviral response is initiated in EC cells following DENV infection.

DENV-2 infection of EC induces STAT1 phosphorylation and total STAT1 protein.
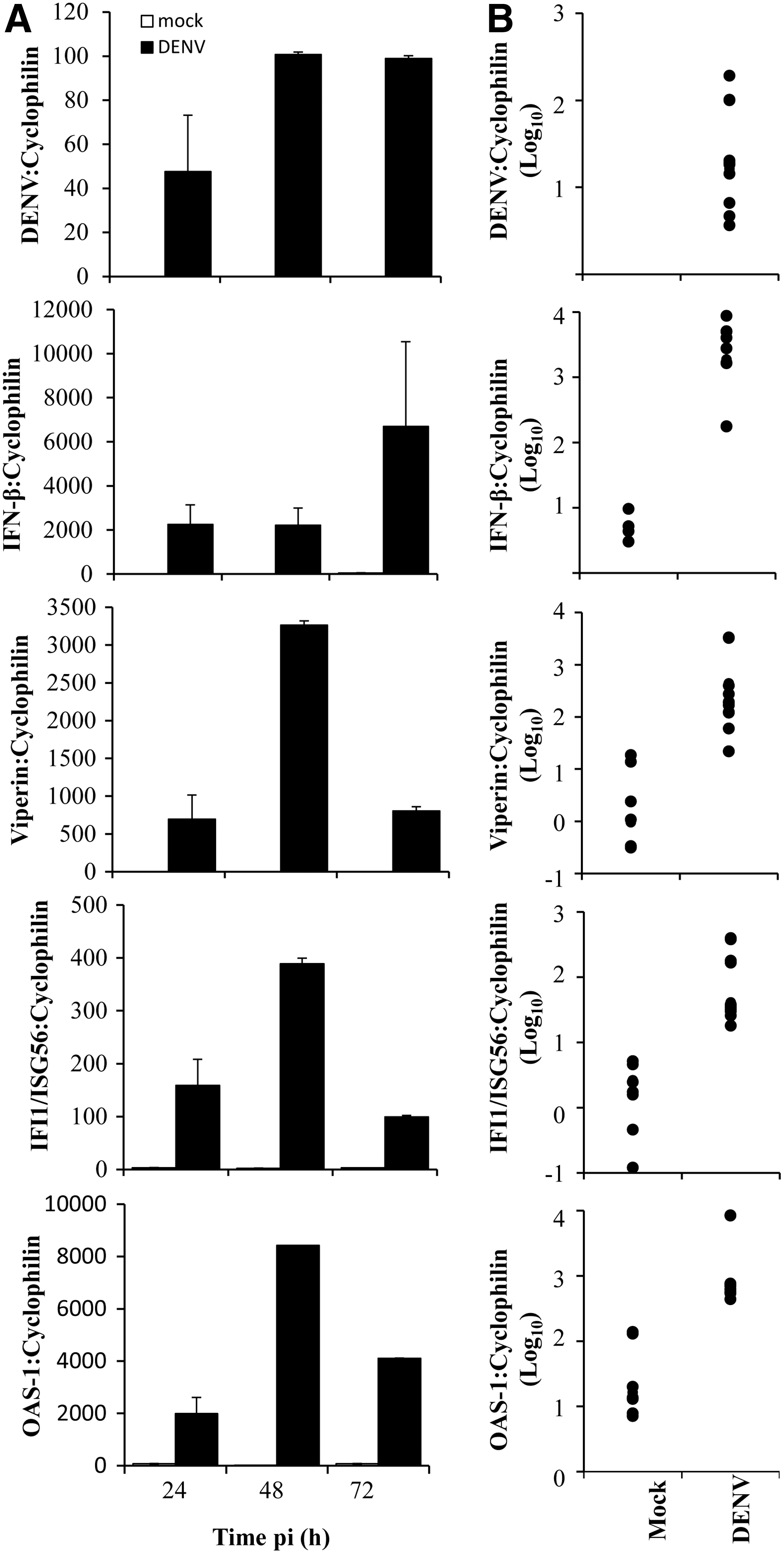
DENV-2 infection of EC induces innate immune responses. EC were mock or DENV-2 infected, RNA extracted, and subjected to RT-PCR. Results were normalized against cyclophilin.
Cells were next infected with DENV-2 and incubated with a neutralizing antibody against IFN-β. At 48 h pi cells were fixed for immunostaining and RNA extracted for RT-PCR. At 48 h pi DENV-2 infection was enhanced in HUVEC incubated with antibody against IFN-β, as measured by production of DENV-2 RNA (Fig. 4A) although production of infectious virus was not significantly different (Fig. 4B). Consistent with this, staining for dsRNA was visualized in increased numbers of HUVEC in cells treated with the IFN-β antibody but the majority of cells still remained uninfected (Fig. 4C). Treatment of cells with a neutralizing antibody against IFN-β also reduced the level of mRNA for OAS-1, IFIT1/ISG56, and viperin but not IFN-β itself (Fig. 4D). Immunostaining of fixed cells for viperin and STAT1-S727 unfortunately was not successful and thus we could not formally demonstrate if the induction of this response was in DENV-2-infected or uninfected bystander cells.

Blocking of IFN-β actions increases DENV-2 infection and reduces mRNAs for interferon-stimulated genes (ISGs). HUVEC were DENV-2 infected and incubated with control or 1,000 U/mL of a neutralizing antibody against IFN-β. At 48 h pi cells were lysed and RNA extracted or cells fixed and immunostained.
We have previously reported that viperin has antiviral actions against DENV (Helbig and others 2013) and thus next investigated the role of viperin in restricting DENV infection by shRNA-mediated knockdown of viperin in EC. HUVEC were transduced with a viperin shRNA lentivirus that has been previously demonstrated to reduce the induction of viperin via IFN-α stimulation by 90%–95% (Helbig and others 2011), a control lentivirus or mock transduced. Cells were allowed to recover and 3 days later challenged with DENV-2. A reduction in viperin mRNA levels was achieved in viperin shRNA compared with control lentivirus transduced cells at day 2 and 3 post transduction (Supplementary Fig. S1; Supplementary Data are available online at
Following DENV-2 infection there was a modest although significant increase in production of infectious virus from viperin shRNA cells at 48 h pi (Fig. 5A). Immunostaining for dsRNA and quantitation by visual cell counting demonstrated an increased number of infected cells in viperin shRNA cultures, although the majority of EC still remained uninfected (Fig. 5B; Table 2). This suggests that viperin can inhibit, but does not completely restrict DENV-2 infection of EC.

Viperin shRNA transduction enhances DENV-2 infection of EC. HUVEC were mock transduced or transduced with a control or viperin shRNA lentivirus. At 72 h post transduction, EC were DENV-2 infected and at 48 h pi
Cell counts were averaged from 5 fields of view from 3 independent HUVEC isolation and lentivirus transduction experiments.
DENV infection of EC induces SK1 activity and alters EC barrier integrity
Our prior studies have identified SK1 as a vital molecule for EC function such as survival and differentiation (Bonder and others 2009; Gamble and others 2009) that we know is also altered during DENV infection (Wati and others 2011; Carr and others 2013). We thus quantified SK1 activity in DENV-infected EC lysates. At 24 h pi, SK1 activity was significantly increased in DENV-2 infected EC, however, by 48 h pi, SK1 activity had normalized and showed no difference between mock and DENV-2 infected EC (Fig. 6).
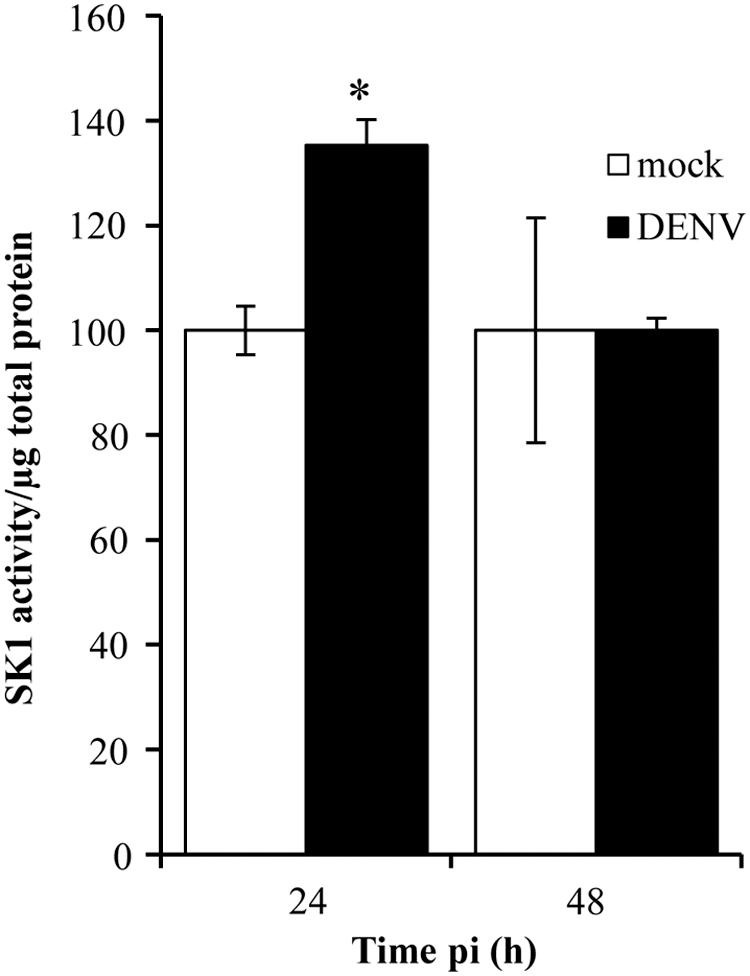
Sphingosine kinase 1 (SK1) activity is increased early following DENV-2 infection of EC. EC were mock or DENV-2 infected and at the indicated time point pi, cells were lysed and SK1 activity quantitated. Results were normalized against total protein and represent mean±SEM from duplicate results from 3 independent donor and infection experiments. *P<0.05, Student's unpaired t-test.
The induction of components of the innate immune response (Fig. 3) and changes in SK1 activity (Fig. 6) suggest the potential for functional changes in DENV-2 infected EC and we therefore next assessed the barrier integrity of DENV-2 infected EC monolayers. HUVEC were mock or DENV-2 infected, then plated in a chamber slide at a density we previously defined to form a monolayer. The cell index, which reflects changes in cell number or function was quantified by real-time measurement of electrical impedence using the x-celligence system. Cell monolayers were viewed by microscopy using the E-view plate system to confirm cell viability and confluence. Results were normalized in each experiment against mock infected controls and demonstrate changes in cell index or barrier integrity with time (Fig. 7).

DENV-2 infection of EC decreases basal barrier integrity in the early hours following infection. EC were mock or DENV-2 infected, replated in E-view plates and incubated in the x-celligence with hourly measurements of barrier integrity. Cells were allowed to recover for 2 h, media changed, and incubation continued for a further 22 h in either
The initial barrier integrity of DENV-infected EC appears lower at 1 h pi, although this is not significant. The spike at 2 h pi reflects a transient change in barrier integrity due to removal of the chamber slide from the incubator to change the media. A significant reduction in cell index, reflecting a decrease in the integrity of the EC barrier, was observed in DENV-infected EC that subsequently recovered to normal within the first 24 h of infection. This effect was observed with either EC cultured in complete HUVEC media (Fig. 7A) or in low serum media [0.1% (v/v) FCS; Fig. 7B]. Consistent with our prior observations (Fig. 1C), in multiple infection experiments from different EC donors there was no evidence of a visual DENV-associated CPE.
EC responses to vasoactive stimuli were assessed at 24 h pi. EC were DENV-2 infected and plated in x-celligence chambers, allowed to recover for 3 h in complete HUVEC media, then serum starved overnight (0.1% FCS, no growth factors). Cultures were then treated with TNF-α or refed complete HUVEC media. Notably, at 24 h pi (time 0 for addition of TNF-α or complete media) the barrier integrity of DENV-EC appears reduced, although this is not significant. At 6–7 h post addition of TNF-α, a greater reduction in EC barrier integrity was observed in DENV-2 compared to mock-infected EC (Fig. 8A). In contrast, restimulation of EC with 20% FCS/growth factors following serum starvation, maintained the barrier integrity of the mock-infected EC monolayer for up to 8 h, but that of DENV-infected EC declined (Fig. 8B).

DENV-2 infection of EC sensitizes EC to stimulation. EC were mock or DENV-2 infected, plated in E-view plates, and allowed to recover for 3 h. Media was changed to 0.1% (v/v) serum media and cells incubated overnight. Media was then replaced with
Together these results suggest time-dependent changes in SK1 activity and functional changes in both basal and growth factor-stimulated EC barrier integrity.
Discussion
Emerging evidence suggests an important role of the endothelium for innate immune responses during viral infections that could also be a potential therapeutic target to prevent tissue and vascular damage (Teijaro and others 2011; Oldstone 2013; Baccala and others 2014; Safronetz and others 2014). The endothelium is a clear target for the pathogenesis of DENV disease. While the degree to which the endothelium contributes to DENV replication is controversial there are well-described functional changes in the endothelium that are linked to severe DENV disease (Basu and Chaturvedi 2008; Dalrymple and Mackow 2012b, 2014). Most studies in primary EC and EC lines demonstrate a low level of EC susceptibility to productive DENV-infection with anywhere from 2% to 30% of EC becoming infected within 1–7 days of infection (Andrews and others 1978; Bunyaratvej and others 1997; Diamond and others 2000a; Huang and others 2000; Warke and others 2003; Peyrefitte and others 2006; Arevalo and others 2009; Liu and others 2009; Zamudio-Meza and others 2009; Vervaeke and others 2013). DENV is reported to utilize heparin sulfate proteoglycansfor entry into a variety of cell types (Chen and others 1997; Hilgard and Stockert 2000; Germi and others 2002; Lin and others 2002; Artpradit and others 2013) including EC (Dalrymple and Mackow 2011; Vervaeke and others 2013) and in the study by Dalrymple and Mackow (2011), DENV-4 infection of EC at MOI of 6 in the absence of heparin resulted in up to 90% of HUVEC DENV-antigen-positive at day 1 pi, as assessed by immunohistochemistry. Similarly, in our study, exclusion of heparin from the cell culture medium increased by 0.5–1.5 log production of infectious virus from DENV-2-infected HUVEC. However, in our experiments, at best only around 15% of the EC population became DENV antigen and dsRNA positive. The study of Vervaeke and others (2013) utilized primary dermal microvascular EC or microvascular EC lines infected with DENV-2 (NGC) and achieved infection of 10%–50% of cells in the absence of heparin that was dependent on MOI, while∼40% of commercially acquired HUVEC were reported to become DENV-2 (16681) infected at day 3–7 pi in the absence of heparin (Raekiansyah and others 2014). Clearly, heparin inhibits DENV-infection of EC and the discrepancies between studies in the actual level of EC susceptibility to DENV-infection may be due to technical factors such as EC senescence (AbuBakar and others 2014), the DENV strain, including potential adaptation to utilize heparin (Lee and others 2006; Artpradit and others 2013; Celis and others 2014) or the presence of DENV NS1 in the inoculums, which can bind to glycosaminoglycans and heparin (Avirutnan and others 2007) and may competitively inhibit DENV infection of EC.
Regardless, EC are susceptible to DENV infection and in our infection system DENV-2 does not spread to infect the majority of cells in culture when using EC from multiple donors, representing 2 different biological sources of EC and including the novel ECFC type, derived from EC progenitors.
We confirmed the induction of IFN-β mRNA in DENV-infected EC as previously reported (Warke and others 2003; Dalrymple and Mackow 2012a; da Conceicao and others 2013). Additionally, we report the induction of OAS-1, IFIT1/ISG56, and viperin mRNA and increased level of both total STAT1 protein and STAT1-S727 and Y701 phosphorylation in DENV-2-infected EC. The observation that total STAT1 protein is increased at 72 h pi in DENV-2-infected EC is interesting and may in part account for the increase seen in STAT1 phosphoprotein. Recent work has demonstrated that a second wave of induction of ISGs is generated through a prolonged production of IFN-β, which could drive an increase in unphosphorylated forms of STAT1, STAT2, and IRF9 (Cheon and others 2013). Similarly, the production of IFN-β, for which we observed mRNA out to at least 72h pi, may be responsible for the increased total STAT1 in our study.
As expected, IFN-β blocking experiments reduced the RNA levels of OAS-1, IFIT1/ISG56, and viperin demonstrating that induction of these genes is dependent on the action of IFN-β. Blocking of IFN-β enhanced DENV RNA production and number of dsRNA-containing cells, but not the amount of infectious virus in cell culture supernatants. This suggests that when IFN-β actions are blocked after infection, production of virus from existing infected cells is unaltered, as expected (Diamond and others 2000b) but additional cells become DENV susceptible and new rounds of infection are increased. These results strongly imply an IFN-β-dependent induction of responses and restriction of infection in bystander cells. Unfortunately, immunostaining for endogenous viperin or STAT-1-S727 in HUVEC could not be achieved to conclusively demonstrate this.
Additionally, lentivirus transduction of EC with shRNA against the ISG viperin also increased DENV infection. This result is similar to our described antiviral role for viperin against DENV-2 infection in macrophages, another important target cell type for DENV infection and pathogenesis, and where similarly a low percentage of cells are DENV-infected, which is associated with induction of viperin, particularly in uninfected bystander cells (Helbig and others 2013). Notably, transduction of viperin shRNA into EC in our study, or blocking with a neutralizing IFN-β antibody did not result in a complete takeover of the EC culture by the DENV.
We demonstrated lower levels of viperin mRNA in viperin shRNA cells prior to and at the time of DENV-infection, have previously validated our viperin shRNA to yield 95% knockdown of IFN-stimulated viperin in Huh7 cells (Helbig and others 2011), and our prior work has shown a high lentivirus transduction rate of EC (Barrett and others 2011). Our results herein, however, suggest that the viperin shRNA knockdown and blocking of IFN-β activity may be incomplete or perhaps overwhelmed by the strong induction of innate responses by DENV infection of EC. Additionally, there are likely to be other antiviral factors that are IFN-β-independent or unaffected by viperin shRNA that can still act to restrict infection in these cells, as suggested by the large number of ISGs that are known to target positive strand RNA viruses in other cell types (Schoggins and others 2014).
Although DENV-infection of EC does not occur in a major fraction of the cell population, the infection clearly still has a significant effect on EC function. We demonstrate an increase in SK1 activity at 24 h pi, a factor that we have previously shown to be an essential regulator of EC survival, growth, and differentiation (Bonder and others 2009; Gamble and others 2009). The increase in SK1 activity in DENV-infected EC may reflect a viral-induced response to prevent premature death of newly infected cells, as has been suggested for the increased SK1 seen early following respiratory syncytial virus and human cytomegalovirus infection in vitro (Monick and others 2004; Machesky and others 2008). Additionally, SK1 has been proposed to play a role in influencing the JAK/STAT pathway following IFN signaling to activate ISGs (Seo and others 2011).
The detected increase in SK1 activity early in DENV infection differs to our prior work showing reduced SK1 activity late in DENV-infected cells, which is mediated via high levels of the DENV 3′UTR of the genome (Wati and others 2011; Carr and others 2013). Thus, the increase in SK1 seen early in DENV-infected HUVEC is mechanistically different to the later decrease in SK1 activity and likely to be driven by antiviral host responses rather than viral replication per se.
Based on our prior work with SK1 and EC, we would expect that elevated levels of SK1 activity would promote EC barrier integrity (Li and others 2008) and sensitize EC to stimulation with inflammatory cytokines (Limaye and others 2009). We assessed this by quantifying real-time changes in EC barrier integrity in vitro. Our data demonstrate significantly different changes in EC barrier integrity in mock and DENV-2-infected EC. Barrier integrity of DENV-2-infected EC was decreased in the early hours following infection, was more pronounced in EC grown in the absence of growth factor supplementation but normalized again by 24 h pi. This suggests an acute response following the insult of a viral infection where the integrity of the EC barrier is compromised. The increased SK1 activity we detected at 24 h pi may subsequently restore normal vascular barrier function.
Additionally, we observed that DENV-infected EC have altered responsiveness to TNF-α and growth factor stimuli, as previously reported (Carr and others 2003; Dewi and others 2004, 2008; Cardier and others 2005) and consistent with elevated SK1 activity at 24 h pi sensitizing cells to the actions of TNF-α (Limaye and others 2009). Some prior studies have suggested that DENV-infected EC do not show altered permeability or responses to stimuli such as TNF-α in vitro (Talavera and others 2004; Liu and others 2009; Raekiansyah and others 2014). These studies, however, utilized single time points at later times pi for analysis of barrier integrity (eg, 7 days pi), and thus may have missed the transient early changes in EC function identified in our study.
The changes in DENV-EC barrier function we have identified in vitro are likely to have functional implications for the endothelium during DENV infection in vivo, in particular for relevant biological stimuli, such as TNF-α, which are known to be elevated in the circulation of DENV-infected patients.
In conclusion, we have presented data demonstrating that DENV-2 can productively infect EC, but infection is restricted and associated with a strong induction of the innate immune response and functional changes in EC. These results support the growing evidence for an important role of DENV-infected EC, not as a major site of viral replication, but as a contributor to the inflammatory response and functional changes in the endothelium that may be associated with severe DENV-disease in vivo.
Footnotes
Acknowledgments
We thank Mrs. Lorena Davies and Julia Zebol for performing the SK1 activity assays. We also thank Dr. Michael Michael, Dr. Karen Humphries, and Ms. Kim McNicholas for access to and assistance with the use of the x-celligence system. This research was supported by funding provided by the Flinders Medical Centre Research Foundation, the National Health and Medical Research Council (NHMRC) of Australia, the Australia-India Strategic Research Fund (J.M.C.), the Fay Fuller Foundation, a NHMRC Senior Research Fellowship (S.M.P.), and a Heart Foundation Fellowship (C.S.B.).
Author Disclosure Statement
No competing financial interests exist.