
Abstract
Plasmacytoid dendritic cells (pDCs) display altered immune-phenotype in multiple sclerosis (MS) patients and are found actively recruited in postmortem MS brain lesions, implying that their immune regulation may represent an important aspect of MS pathogenesis. Because of the reported Toll-like receptor 7 (TLR7) implication in autoimmunity, in this study we characterized how IFN-β therapy impacts on pDC activation to TLR7 triggering in MS patients, aspect only poorly investigated so far. In vivo IFN-β administration regulates pDC functions in TLR7-treated peripheral blood mononuclear cell (PBMC) cultures differently from what is observed in isolated cells, suggesting that IFN-β may activate inhibitory mechanisms in MS peripheral blood involved in turning off pDC response to dampen the ongoing inflammation. Indeed, IL-10, a key regulatory cytokine found increased upon TLR7 stimulation in in vivo IFN-β-exposed PBMCs, directly reduced pDC-mediated IFN-α production. IFN-β therapy also shaped T-cell responses by decreasing TLR7-induced pDC maturation and inducing T-cell inhibitory molecules. Accordingly, raised pDC-induced IL-27 and decreased IL-23 expression, together with high IL-10 level, contribute to inhibit Th17 cell differentiation. Our study uncovered a role for IFN-β in the regulation of TLR7-mediated pDC responses in MS toward an anti-inflammatory phenotype opening new opportunities to better understand mechanisms of action of this drug in controlling MS immunopathogenesis.
Introduction
M
Plasmacytoid dendritic cells (pDCs) have been studied in MS for their ability to stimulate or inhibit effector T cells, assumed to initiate and perpetuate the autoimmune reactions involved in MS pathogenesis. Indeed, pDCs are present in the cerebrospinal fluid (CSF) of MS patients, compartment where their concentration is increased during exacerbation of disease (Longhini and others 2011). We also demonstrated that these cells could be found in leptomeninges and demyelinating lesions of MS-affected individuals (Lande and others 2008). Furthermore, dysregulated pDC functions and responses have been described contributing to impairment of immune-regulatory mechanisms in MS patients (Stasiolek and others 2006; Sanna and others 2008; Bayas and others 2009; Schwab and others 2010; Hirotani and others 2012).
pDCs have been initially identified for their intriguing ability to produce more type I IFNs than any other cell type in response to viral stimulation (Swiecki and Colonna 2010). Multiple receptors have evolved in mammals for detecting pathogen-associated molecular patterns, but pDCs are unique also for their selective expression of Toll-like receptor (TLR) 7 and TLR9, endosome-lysosome resident molecules able to specifically sense microbial or self-derived nucleic acids (Gilliet and others 2008). TLR7 recognizes single-stranded RNA derived from viruses or synthetic imidazoquinoline-like compounds, while ligand of TLR9 is double-stranded DNA and, in particular, CpG-rich motifs found in viral and bacterial DNA.
Several lines of evidence suggest how microbial factors might contribute to the etiopathogenesis of MS, with Epstein-Barr virus (EBV) considered one of the major candidates (Ascherio 2008). Evidence of EBV infection was found in the majority of brain-infiltrating B and plasma cells with ectopic B-cell follicles forming in the cerebral meninges identified as major sites of EBV persistence (Serafini and others 2007, 2010). pDCs were detected in the close proximity of EBV-infected sites in the highly inflamed MS brains (Serafini and others 2007). Consistent with these data and with the fact that pDC-driven antiviral activity can have a role in the pathogenesis of MS collaborating in the dysregulation of immune responses, we recently demonstrated that pDCs are a novel target of EBV infection and that this virus can subvert pDC functions to block T cell-specific responses and to favor its own persistence in the host (Severa and others 2012).
To improve the current therapeutic strategies in treating MS, nowadays there is increasing interest in the usage of antiviral therapies in autoimmune disorders and in particular in MS (Dreyfus 2011). In this context, IFN-β, currently one of the first-line disease-modifying therapies more frequently used for relapsing remitting MS (RRMS), can have a very relevant place (Axtell and Raman 2012). Indeed, even if it is believed that IFN-β efficacy mainly depends on its variegate immune-regulatory activities (Severa and others 2014), it is also very much likely that the antiviral action of this cytokine, belonging to the type I IFN family discovered more than 50 years ago for the ability to interfere with and block viral replication (Borden and others 2007), might be relevant in the treatment of autoimmune diseases.
To assess whether IFN-β therapy would modulate pDC-driven inflammatory processes, in the past we studied the impact of in vivo IFN-β treatment on TLR9-induced pDC responses in MS patients (Lande and others 2008). We found that following TLR9 stimulation pDCs isolated from IFN-β-treated MS patients display reduced maturation and capacity to produce IFN-α compared with their counterpart purified from the same individuals before the beginning of IFN-β administration, resulting in a decreased ability to induce allogeneic T cell responses. These results are in line with what was observed recently by Balashov and others (2010) showing that in patients undergoing IFN-β therapy the reduction in proinflammatory cytokine release by TLR9-stimulated pDCs was due to a significant impairment in TLR9 processing, a N-terminal cleavage of the protein necessary to make it functional and activate intracellular signaling.
Based on this background, we sought to investigate whether the modulation of pDC functions by IFN-β could be extended also to TLR7-induced responses. Our interest on TLR7 relies on multiple aspects. First of all, this receptor has been implicated at different levels in autoimmunity with polymorphisms in TLR7 gene shown to have a role in time to disease progression in individuals affected by MS (Enevold and others 2010) but also in predisposition to systemic lupus erithematosus in Asian population (Kawasaki and others 2011). Second, in the past we showed that TLR7 transcription is IFN-β-inducible (Severa and others 2007) and our recently published data demonstrate how TLR7-induced B-cell differentiation into immunoglobulin (Ig)-secreting cells is impaired in MS patients, accordingly to a specific deficiency in TLR7 gene expression, dysregulation that was corrected following IFN-β administration (Giacomini and others 2013).
In this study we observed that in vivo IFN-β treatment modifies the ability of pDCs derived from MS patients to mature and secrete type I IFN when stimulated by TLR7 in the context of unseparated peripheral blood mononuclear cells (PBMCs), differently to that found in purified pDCs.
Our experimental approach, based on cultures of whole PBMCs, allowed the study of pDCs in a physiological setting more closely resembling that found in the blood of MS patients and led to the dissection of immune-regulatory aspects, including the crosstalk among different cell types either via cell–cell contact or as a result of bystander responses, which cannot be taken into account when studying isolated cells.
By using this experimental setting, we also observed that IFN-β therapy changes the Th17-skewing attitude of pDCs by modifying their phenotype and their cytokine production, inducing IL-27 and repressing IL-23 toward an inhibition of Th17 differentiation.
Materials and Methods
Patients
Fifteen patients with definite RRMS (median age 38.9, range 26–53 years) according to the revised McDonald's criteria (Polman and others 2005) were enrolled at S. Andrea Hospital, following the standard procedures of the outpatients' service of the MS Center. Patients were 65% women and 35% men, respecting the female-to-male ratio of MS that in Europe varies from 1.5 to 3. Ethics Committee of S. Andrea Hospital approved the study (CE 204/10) and all the subjects involved gave written informed consent.
The main demographic and clinical characteristics of recruited patients are shown in Table 1. The Expanded Disability Status Scale scores ranged from 0 to 3.0 (mean 1.6) and disease duration was from 1 to 17 years (mean 7.5). To be included in the study, patients had to be free of immunosuppressive therapies for at least 3 months and before the enrollment a minimum of a 6-month-period of washout from any other immunomodulatory drug was considered. Magnetic resonance imaging was performed for each patient within 30 days from sampling.
All patients had relapsing-remitting MS. The reported characteristics are referred to patients before the beginning of IFN-β therapy.
EDSS, expanded disability status scale; MRI Gd enhancement, nuclear magnetic resonance imaging gadolinium enhancement; MS, multiple sclerosis.
Patients were longitudinally followed right before (T0) and 1 month after (T1) the beginning of IFN-β treatment (recombinant IFN-β1b in the formulation of Betaferon™ 250 μg subcutaneously, every other day; Bayer). We chose to study 1 month post the beginning of therapy to make sure to analyze a time window in which anti-IFN-neutralizing antibodies (Abs) were not produced yet and, thus, could not mask or alter patients' responsiveness. A posteriori, however, 3 of the 15 patients analyzed were clinically defined as nonresponders and discontinued IFN-β administration. Even so, no significant difference in their responsiveness was observed in our analyses at T0 and T1 in respect to the responder individuals.
Cell isolation, culture, and stimulation
Peripheral blood (40–50 mL) was collected from MS patients and processed within 16 h from sampling. PBMCs and pDCs were obtained as previously described (Lande and others 2008).
For specific cell-type depletion, PBMCs were subjected to positive sorting using anti-CD14 conjugated magnetic microbeads (Miltenyi Biotec) to remove monocytes or anti-BDCA4 beads to deplete pDCs from whole PBMCs.
pDCs (0.5×106/mL) or PBMCs (1×106/mL) were stimulated with 5 μM of the synthetic specific TLR7 agonist Imiquimod-3M001 (a kind gift of Dr. Mark Tomai; 3M pharmaceuticals). Recombinant IL-10 (PeproTech) was used at 25 ng/mL.
Flow cytometry analysis
To evaluate the percentage of pDCs, PBMCs (3×105) were stained with the Lineage cocktail 1 (BD Pharmingen) containing Abs that, in combination, stain lymphocytes, monocytes, eosinophils, and neutrophils. In the Lineage negative fraction of a lympho-mono gate, we considered as pDCs the cells triple positive for IL3Ra (CD123), HLA-DR (BD Pharmingen), and BDCA2 (Miltenyi Biotec). The full gating strategy is shown in Supplementary Fig. S1 (Supplementary Data are available online at
The expression on the pDC surface of CD86 (BD Pharmingen) or inducible T cell co-stimulator ligand (ICOS-L), B7-H1, Ig-like transcript 7 (ILT7), Fcɛ-RIγ (CD23) and Fcγ-RIIα (CD32) (eBioscience) was evaluated in the Lin−CD123+HLA-DR+BDCA2+ gate by running cells on a FACSCanto (BD Pharmingen) and analyzing data by FlowJo software (TreeStar, Inc.).
Monoclonal Abs and IgG1, IgG2a control Abs (BD Pharmingen) were used conjugated with FITC, PE, PERcP, APC, or APC-Cy7 as needed.
Differentiation of Th17 cells
Total or pDC-depleted PBMCs (1×106/mL) from 4 therapy-free MS patients before or after in vivo IFN-β therapy were cultured for 24 h with 5 μM TLR7 ligand 3M001. After 24 h, the cultures were stimulated with anti-CD3-CD28 beads (Invitrogen, Life Technologies) (1 bead: 2×105 PBMCs) for additional 5 days. Then, cells were treated for 4 h with phorbol 12-myristate 13-acetate (40 ng/mL) and ionomycin (1 μg/mL) (Sigma Aldrich) and intracellular staining for IL-17A (BD Pharmingen) was performed by using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences) according to the protocol provided. Intracellular IL-17A expression was evaluated by flow cytometry in gated CD4+ T cells. In culture supernatants IL-17A production was also tested by ELISA (R&D system) and tumor necrosis factor (TNF)-α and IL-10 by using the Th-1/Th-2 Cytometric Bead Array kit (BD Pharmingen).
Positive control cultures for Th17 cell differentiation was set up by in vitro treating PBMCs for 5 days in the presence of anti-CD3-CD28 beads (Invitrogen, Life Technologies) with a cytokine cocktail of recombinant IL-1β (10 ng/mL), IL-6 (20 ng/mL), IL-23 (100 ng/mL), and TGF-β (1 ng/mL) as previously described (Volpe and others 2009). All recombinant cytokines were purchased from Peprotech.
Cord blood-derived naïve allogeneic CD4+ T cells were purified by indirect magnetic sorting with CD4+ T cell isolation kit (Miltenyi Biotech). pDCs, isolated from MS patients at T0 and T1, were treated for 24 h with 5 μM TLR7 ligand 3M001 and then cocultured with naïve T cells at 1:3 T cell/pDC ratio using 3×104 T cells for additional 5 days. IL-17A production was tested in coculture supernatant by ELISA (R&D system).
ELISA
IFN-α kit was purchased from PBL Biomedical Laboratories and IL-10 kit from Bender MedSystems.
RNA purification and real-time reverse transcription polymerase chain reaction
DNase-I-treated total RNA was purified from pDCs isolated from PBMCs of 5 MS patients following a procedure already described (Severa and others 2012). Quantitative polymerase chain reaction (PCR) assays were performed on a LightCycler Instrument (Roche Diagnostics GmbH). Sample values were normalized by calculating the relative quantity of each mRNA to that of gliceraldeide-3-fosfato deidrogenasi (GAPDH) using the formula 2−ΔCt and expressed as mean±SD.
Primer pairs for GAPDH, TLR7, IFN-regulatory factor (IRF) 5, IRF7, IL27-EBI3, IL27-p28, and IL23-p19 transcripts were as previously described (Coccia and others 2004; Remoli and others 2007; Severa and others 2007). The other primers used in this study were as follows:
Myeloid differentiation primary response 88 (MyD88)_forward: 5′-TGGGACCCAGCATTGAGGA-3′
MyD88_reverse: 5′-CGCTGGGGCAATAGCAGA-3′
Suppressor of cytokine signaling 1 (SOCS1)_forward: 5′-AACTGCTTTTTCGCCCTTA-3′
SOCS1_reverse: 5′-GCCACGTAGTGCTCCA-3′
Toll interacting protein (Tollip) _forward: 5′-TCCATTGGGAATGAAGGTGT-3′
Tollip _reverse: 5′-TCTGAGTGTGGCTTGTGGTC-3′
Statistical analysis
Statistical significance of differences was determined for pair data by two-tailed Student's t-test (up to 5 MS patients) or by Wilcoxon signed-rank test (15 MS patients) (P≤0.05 was considered significant).
Results
TLR7-driven IFN-α production in pDCs of MS patients undergoing IFN-β therapy
Over the past years, the response of MS-derived pDCs to TLR9 stimulation was characterized at different levels by others and us (Stasiolek and others 2006; Lande and others 2008; Bayas and others 2009; Balashov and others 2010; Schwab and others 2010; Hirotani and others 2012). Because of the reported TLR7 implications in autoimmunity, in this study we evaluated whether the ability of pDCs to respond to TLR7 triggering could be regulated in MS patients by IFN-β therapy (IFN-β-1b; Betaferon), aspect that has been only poorly investigated so far (Derkow and others 2013).
As a first step we characterized the regulation of IFN-α production, key feature of pDCs. pDCs were purified from PBMCs of RRMS patients longitudinally studied before (T0) and 1 month after (T1) the beginning of IFN-β therapy. Cells were then cultured for 24 h with a specific synthetic TLR7 ligand, Imiquimod-3M001. In these experimental settings, in vivo IFN-β-exposed pDCs produced a much higher level of IFN-α than pDCs from therapy-naïve patients (Fig. 1).
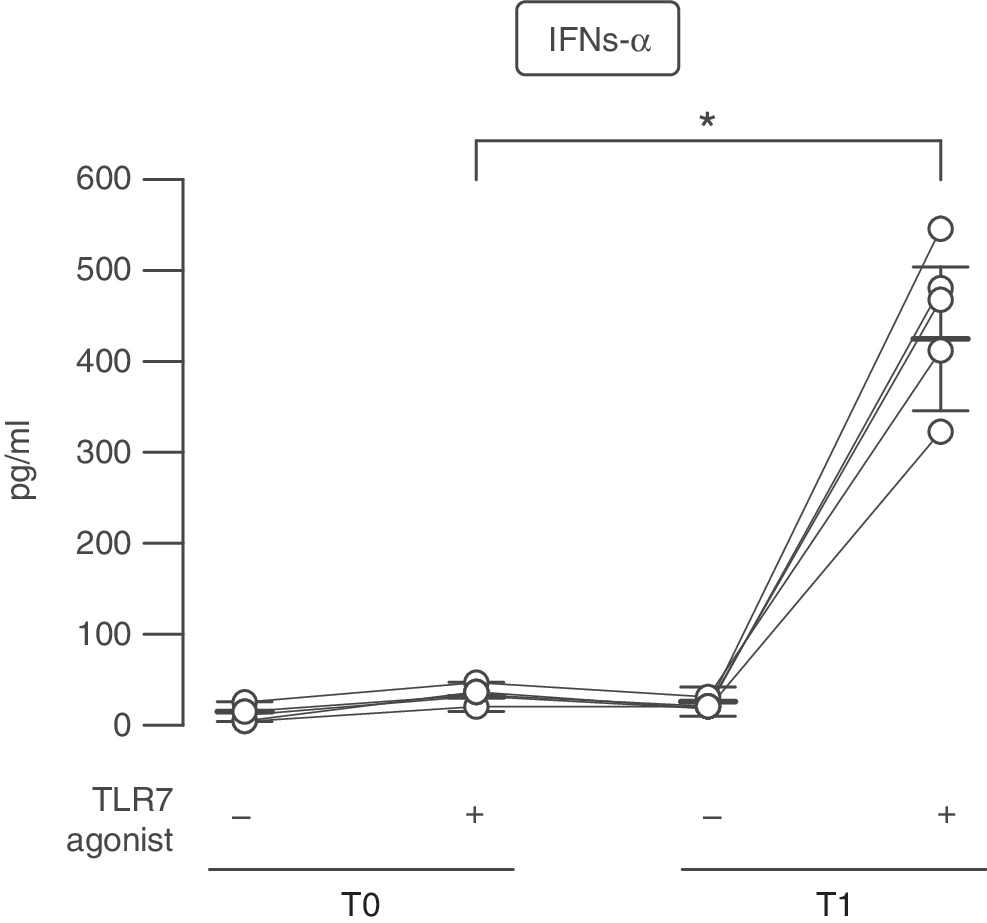
Higher Toll-like receptor 7 (TLR7)-stimulated IFN-α production in plasmacytoid dendritic cells (pDCs) of IFN-β-treated multiple sclerosis (MS) patients. IFN-α production was measured by ELISA in culture supernatants of pDCs derived from 5 MS patients before (T0) and after 1 month (T1) of IFN-β therapy and cultured for 24 h with or without the TLR7 ligand 3M001. Interindividual responsiveness of patients is shown as scatter plot. Data are depicted as mean±SEM. *P=0.025.
These findings well correlate with the transcriptional profile of several components of TLR intracellular signaling evaluated in total RNAs of pDCs derived from 5 MS patients before and after IFN-β therapy. TLR7 associates with the adaptor molecule MyD88, which is responsible for initiation of down-stream intracellular pathways leading to activation of the transcription factors NF-κB, IRF5, and IRF7. Then, their nuclear translocation leads to type I IFN production (Gilliet and others 2008). Real-time PCR analysis showed a significant increase in the level of TLR7, MyD88, IRF7 and IRF5 gene expression in MS-derived pDCs isolated upon IFN-β administration (Fig. 2A).
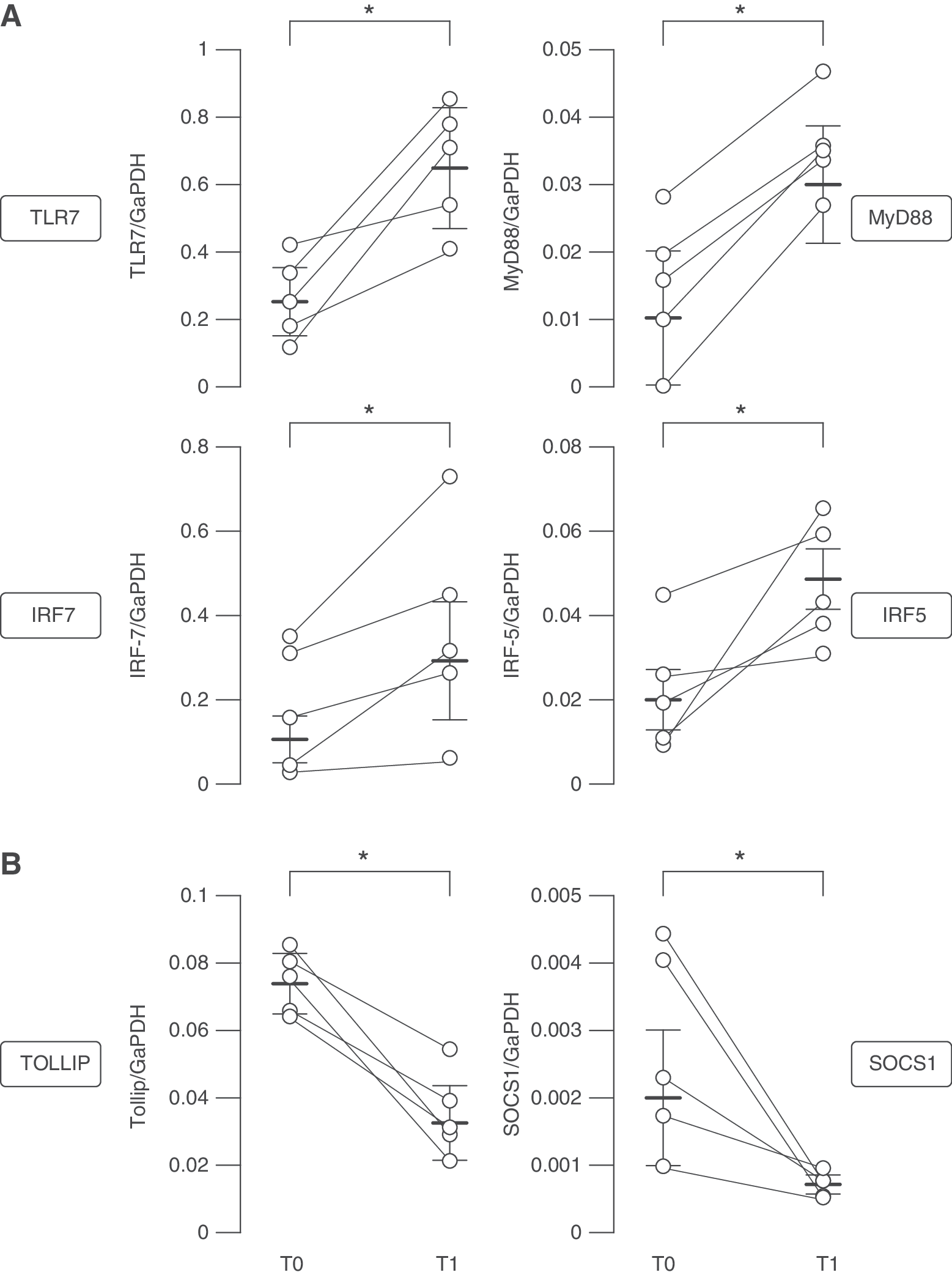
Transcriptional analysis of factors involved in TLR and type I IFN pathway in pDCs of MS patients before and after IFN-β therapy. Total RNA was prepared from freshly isolated pDCs of 5 MS patients collected at T0 and T1. Interpatient variability in ex vivo expression of TLR7, MyD88, IRF7, IRF5
Together with positive regulators of TLR signaling, in recent years a panoply of endogenous TLR inhibitors was identified. Some of these inhibitory molecules, such as Tollip, are expressed at baseline and act to suppress TLR signaling constitutively (Zhang and Ghosh 2002). Other inhibitors are, conversely, themselves induced by TLR stimulation and mediate feedback suppression of TLR response, like SOCS members and SOCS1 in particular (Yoshimura and others 2007). Both Tollip and SOCS1 have been tightly linked to the negative regulation of TLR-induced type I IFN production. In accordance with an increased ability of in vivo IFN-β-conditioned MS pDCs to produce IFN-α upon TLR7 treatment, we found both Tollip and SOCS1 mRNAs strongly down-regulated in pDCs following IFN-β therapy (Fig. 2B).
Different pDC-mediated IFN-α regulation in PBMCs of IFN-β-treated MS patients
While studying TLR7-driven IFN-α production of MS patients-derived PBMCs, an interesting picture emerged. Upon viral or TLR7/9 stimulation, the main producers of type I IFN in PBMCs are pDCs (Coccia and others 2004; Severa and others 2012), as also demonstrated by pDC depletion strategy [data not shown and (Severa and others 2012)]. However, TLR7-stimulated PBMCs derived from IFN-β-treated MS patients showed a strong impairment in IFN-α release, revealing an opposite trend to what found in isolated pDCs (Fig. 3A).
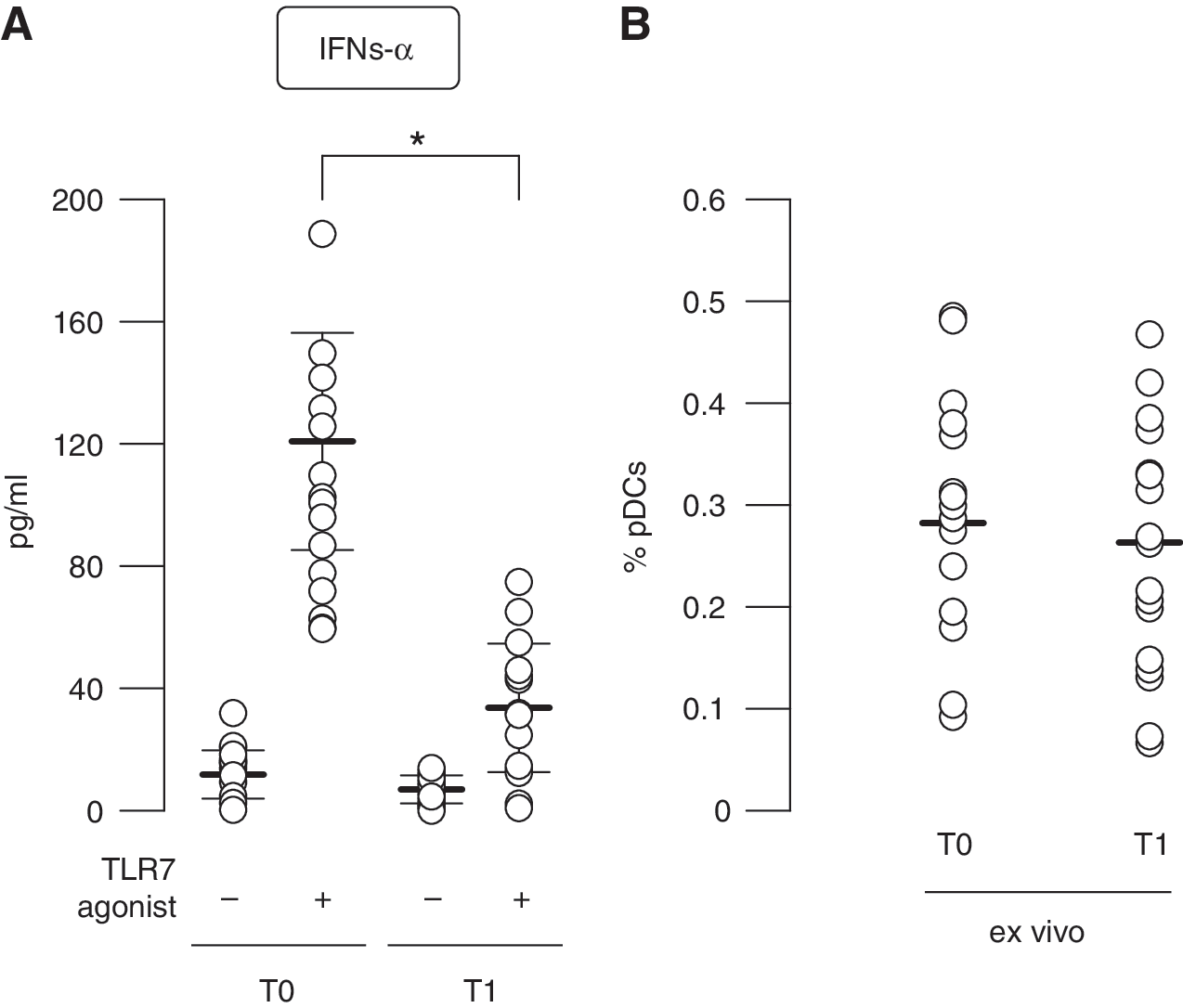
Decrease in pDC-mediated TLR7-induced IFN-α production in the context of peripheral blood mononuclear cell (PBMC) cultures derived from IFN-β-treated MS patients. PBMCs obtained from 15 MS-affected individuals collected at T0 and T1 of IFN-β therapy were cultured for 24 h with or without the TLR7 ligand 3M001.
This was not due to a difference in the percentage of circulating pDCs, since a comparable frequency of this cell type was observed in the blood of MS patients before and after IFN-β therapy (Fig. 3B), as also previously reported by others and us studying diverse IFN-β pharmacological formulations (Lopez and others 2006; Lande and others 2008).
Mechanisms regulating TLR7-driven type I IFN production in pDCs of MS patients undergoing IFN-β therapy
The amplitude of type I IFN responses to TLR ligands is regulated in pDCs by several activating or inhibitory surface receptors that deliver intracellular signals directly or through adapter molecules containing intracellular tyrosine-based activation or inhibitory motifs (Swiecki and Colonna 2010). These receptors include blood dendritic cell antigen-2 (BDCA-2, also called CD303) and ILT7, both complexed with Fcɛ-RIγ, and Fcγ-RIIα (Bave and others 2003; Cao and others 2006, 2007). To identify the mechanisms controlling pDC-driven IFN-α production in PBMCs of IFN-β-treated MS patients, at first we monitored the expression of the aforementioned molecules on pDC surface before and after IFN-β therapy. However, flow cytometry analysis did not highlight any significant modulation of ILT7, Fcɛ-RIγ, or Fcγ-RIIα on MS pDCs by in vivo IFN-β treatment (Fig. 4).
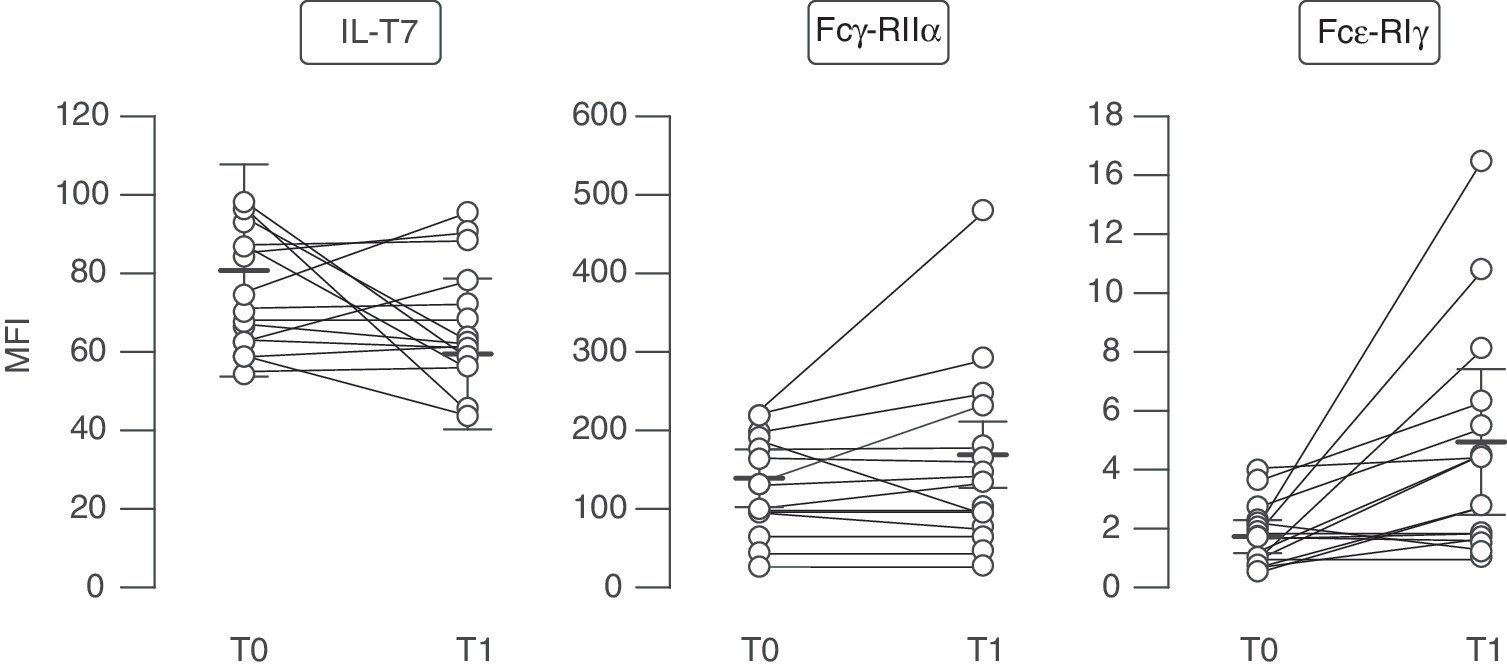
Unchanged expression of different molecules known to regulate IFN-α production in pDCs of IFN-β-treated MS patients. Ex vivo expression of ILT7, Fcγ-RIIα, and Fcɛ-RIγ was analyzed in the Lin−CD123+HLA-DR+BDCA2+-gated pDCs of 15 different MS patients collected at T0 and T1. Data are represented as MFI (mean fluorescence intensity) values±SEM of these molecules.
Since the reduction in TLR7-stimulated type I IFN release is not observed in isolated pDCs but only in the physiological context of mixed-cell populations of PBMCs, these divergent results could be due to the induction of IFN-β-mediated immune-regulatory networks influencing pDC differentiation and activation.
Cytokine production is stringently regulated by bystander responses in immunity and autoimmunity. In particular, IFN-α release by pDCs might be directly inhibited by TNF-α (Palucka and others 2005) or IL-10 (Duramad and others 2003; Murray 2006). While no significant difference was found for TNF-α production (Supplementary Fig. S2A), IL-10 level was found strongly induced upon TLR7 triggering in culture supernatants of PBMCs derived from MS patients undergoing IFN-β therapy (Fig. 5A). Thus, these data suggest that the increased IFN-β-induced IL-10 found in MS PBMCs might be responsible for the negative feedback loop leading to the reduction of pDC-mediated IFN-α production upon TLR7 triggering.

Regulatory role of TLR7-mediated IL-10 production in PBMCs of IFN-β-treated MS patients.
Among the different immune cells, B cells and monocytes are considered main producers of IL-10. While B cells constitutively express TLR7, in steady-state conditions monocytes mainly express TLR8 and only low level of TLR7. However, we recently reported that IFN-β therapy specifically enhances TLR7 gene expression in MS patient-derived monocytes (Giacomini and others 2013). Thus, we depleted both B cells and monocytes from PBMCs isolated from IFN-β-treated patients and measured IL-10 release in TLR7-stimulated cultures. In this experimental setting we found a significantly reduced IL-10 production in the absence of monocytes (Fig. 5B), while B-cell depletion revealed for them only a marginal role in this context (Supplementary Fig. S2B).
To verify our hypothesis that IFN-β-driven IL-10 release mediated by monocytes could inhibit IFN-α production in MS pDCs, we treated isolated pDCs with recombinant IL-10 in the presence or absence of TLR7 agonist (Fig. 5C). This setting clearly demonstrated that IL-10 directly targets TLR7-induced IFN-α production, as also shown for TLR9 stimulation (Duramad and others 2003), unveiling a novel immune-modulatory role for IFN-β therapy as regulator of bystander responses targeting pDC functions in MS patients toward a more anti-inflammatory milieu.
Regulation of pDC immune-phenotype in IFN-β-treated MS patients
In response to inflammatory stimuli, pDCs undergo maturation expressing co-stimulatory molecules, such as CD80 and CD86, and producing proinflammatory cytokines, instructing multiple T-cell functions.
PBMCs isolated from MS patients before and after therapy were treated for 2 days with the TLR7 agonist to reach a full maturation. Flow cytometry analysis revealed that the surface expression of the co-stimulatory molecule CD86 was nicely induced on pDCs from therapy-naïve patients by TLR7 stimulation compared with untreated cultures (Fig. 6A, B). Conversely, the capacity of TLR7-treated pDCs to undergo maturation was impaired upon IFN-β therapy. In addition, in vivo IFN-β-conditioned pDCs display an enhanced expression of B7-H1 (also called programmed death-1 ligand, PD-L1) and ICOS-L, two members of the B7 family of immune-regulatory ligands that deliver co-inhibitory signals to T cells to suppress their proliferation (Collins and others 2005) (Fig. 6A, B).
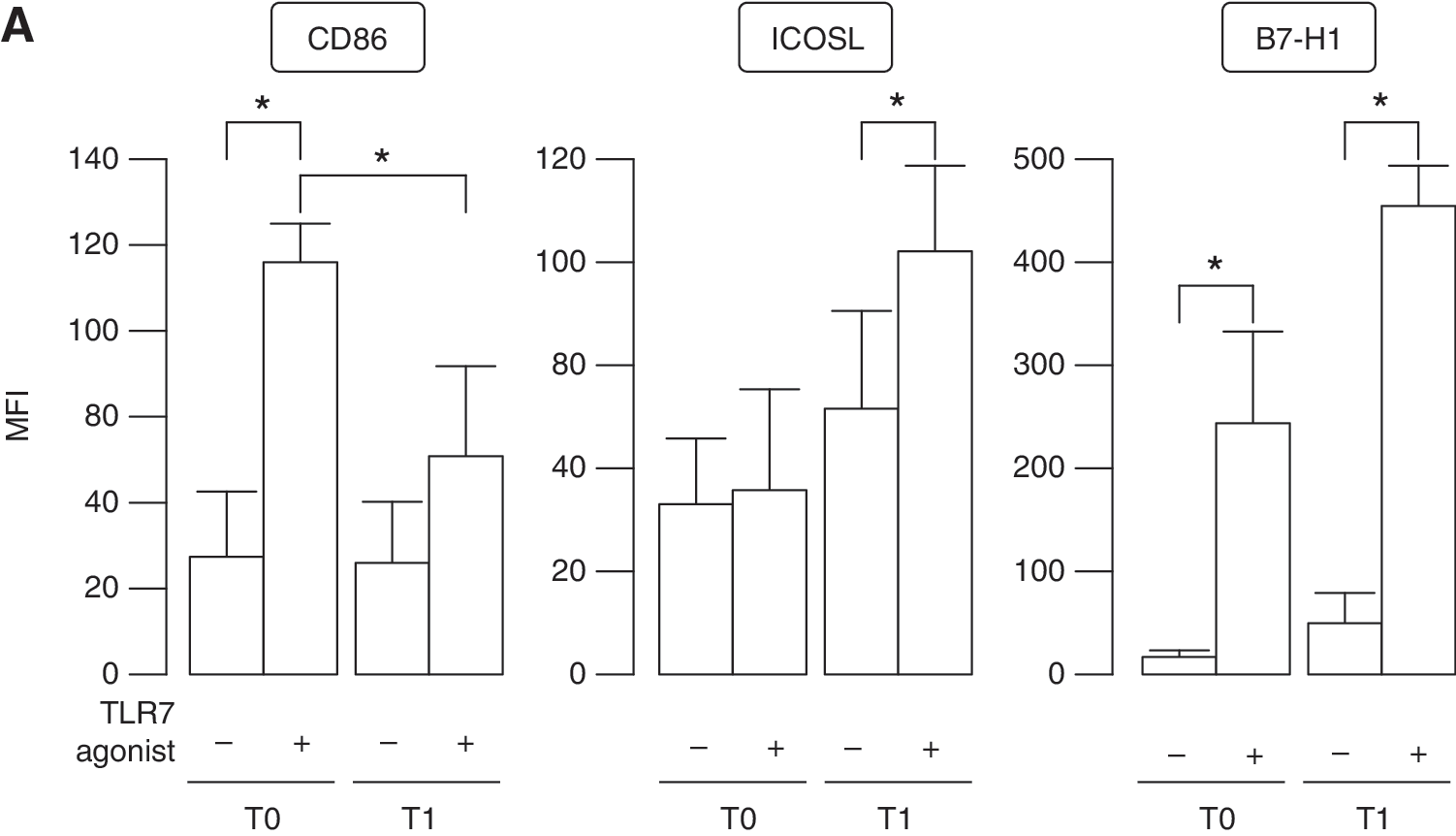
TLR7-regulated phenotype and function of pDCs from IFN-β-treated MS patients.
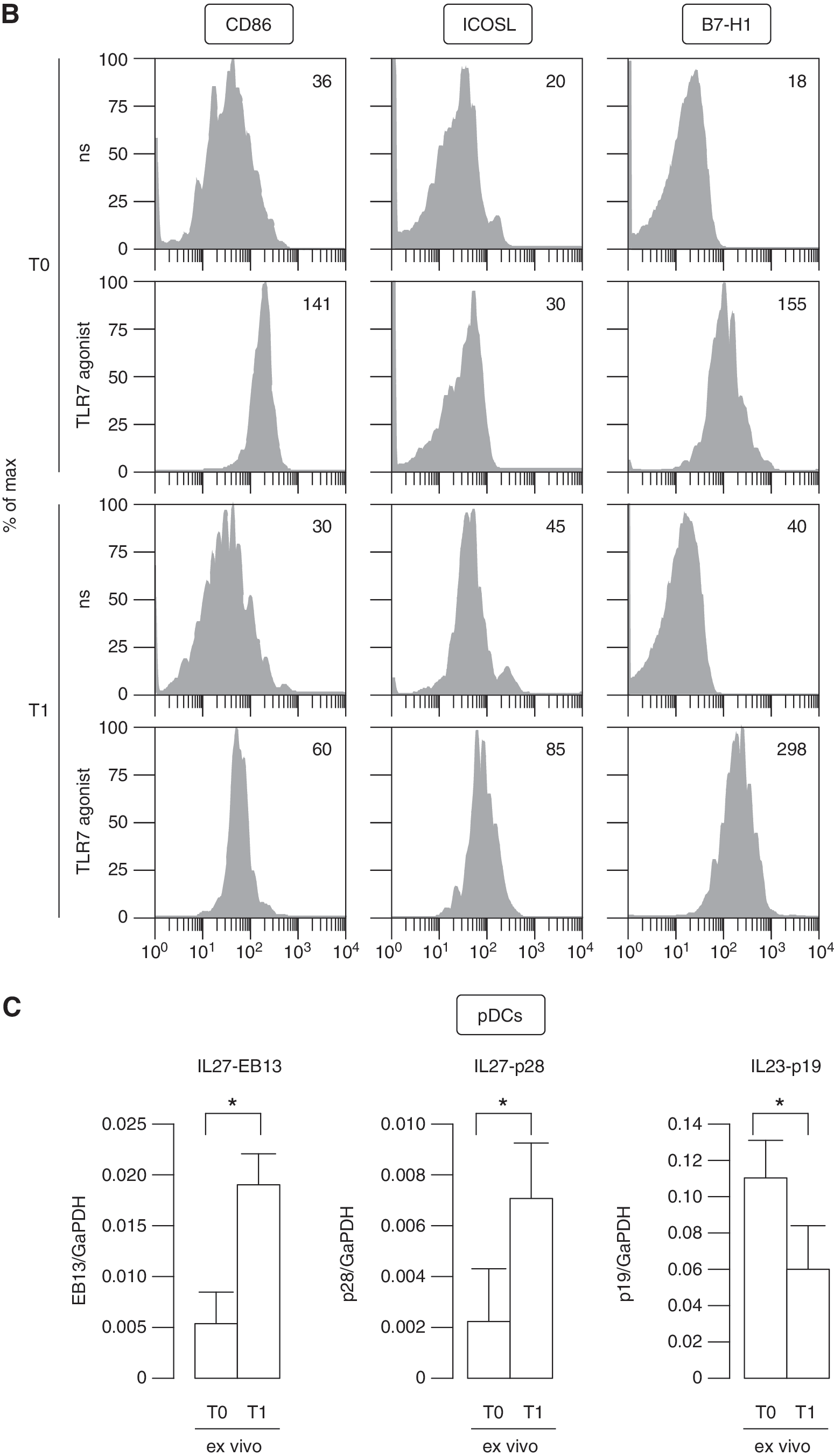
In particular, TLR7 treatment did not directly modulate ICOS-L on pDCs of therapy-free patients, whereas its expression was upregulated by IFN-β administration and further enhanced in the presence of TLR7 triggering. B7-H1 expression followed the same trend than that observed for ICOS-L, even if it was also strongly upregulated by TLR7 treatment in PBMCs from therapy-naïve patients.
Shaping of Th17 response by TLR7-induced pDCs derived from MS patients undergoing IFN-β therapy
In a scenario in which IFN-β therapy reduced the maturation but concomitantly induced upregulation of both the T-cell inhibitory markers ICOS-L and B7-H1 on pDCs of MS patients in response to TLR7 triggering, it is conceivable to assume that the IFN-β-induced phenotype of pDCs might shape the T-cell responses and, in particular, the differentiation of IL-17 producing T cells (Th17), crucial inducers of tissue inflammation and players of organ-specific autoimmunity, including MS (Sallusto and Lanzavecchia 2009).
Since a specific cytokine network is required for the regulation of Th17 responses (Chen and O'Shea 2008; Volpe and others 2009; Segura and others 2013), we investigated the expression of negative, IL-10 and IL-27, and positive, IL-23, regulators of Th17 development. In particular, together with the strong induction of IL-10 that we detected in TLR7-stimulated PBMCs from IFN-β-treated MS patients (Fig. 5A), freshly isolated pDCs from these individuals induced the transcription of both IL27-p28 and -EBI3 subunits (Carl and Bai 2008) as detected by real time reverse transcription polymerase chain reaction (RT-PCR) (Fig. 6C). Conversely, it was very interesting to find that the inducible p19 subunit of IL-23, which contributes to drive Th17 differentiation, was strongly decreased in in vivo IFN-β-conditioned pDCs. Hence, our findings highlighted that pDCs, in the presence of IFN-β, may modify the cytokine milieu producing IL-27 and repressing IL-23 toward an inhibition of Th17 differentiation, as it was also previously reported for other cell types, including monocyte-derived DCs (Ramgolam and others 2009; Zhang and others 2009).
To prove the involvement of pDCs in Th17 expansion, we compared TLR7 responses of whole PBMCs to that of pDC-depleted PBMCs from 4 MS patients at T0 and T1. After 24 h of cultures, T-cell proliferation was induced with anti-CD3/CD28 stimulation for other 5 days and then intracellular production of IL-17A evaluated by flow cytometry on CD4+ T cells. Our data showed that the IL-17A expression and Th17 expansion observed in TLR7-treated MS PBMCs was strongly reduced in the presence of in vivo IFN-β treatment (Fig. 7A–C), as also previously reported (Ramgolam and others 2009; Zhang and others 2009). However, our experimental setting further proved that pDCs might be key modulators of Th17 responses, since in the absence of this cell type in therapy-free MS patients there is a strong reduction in the percentage of IL-17A-producing cells.
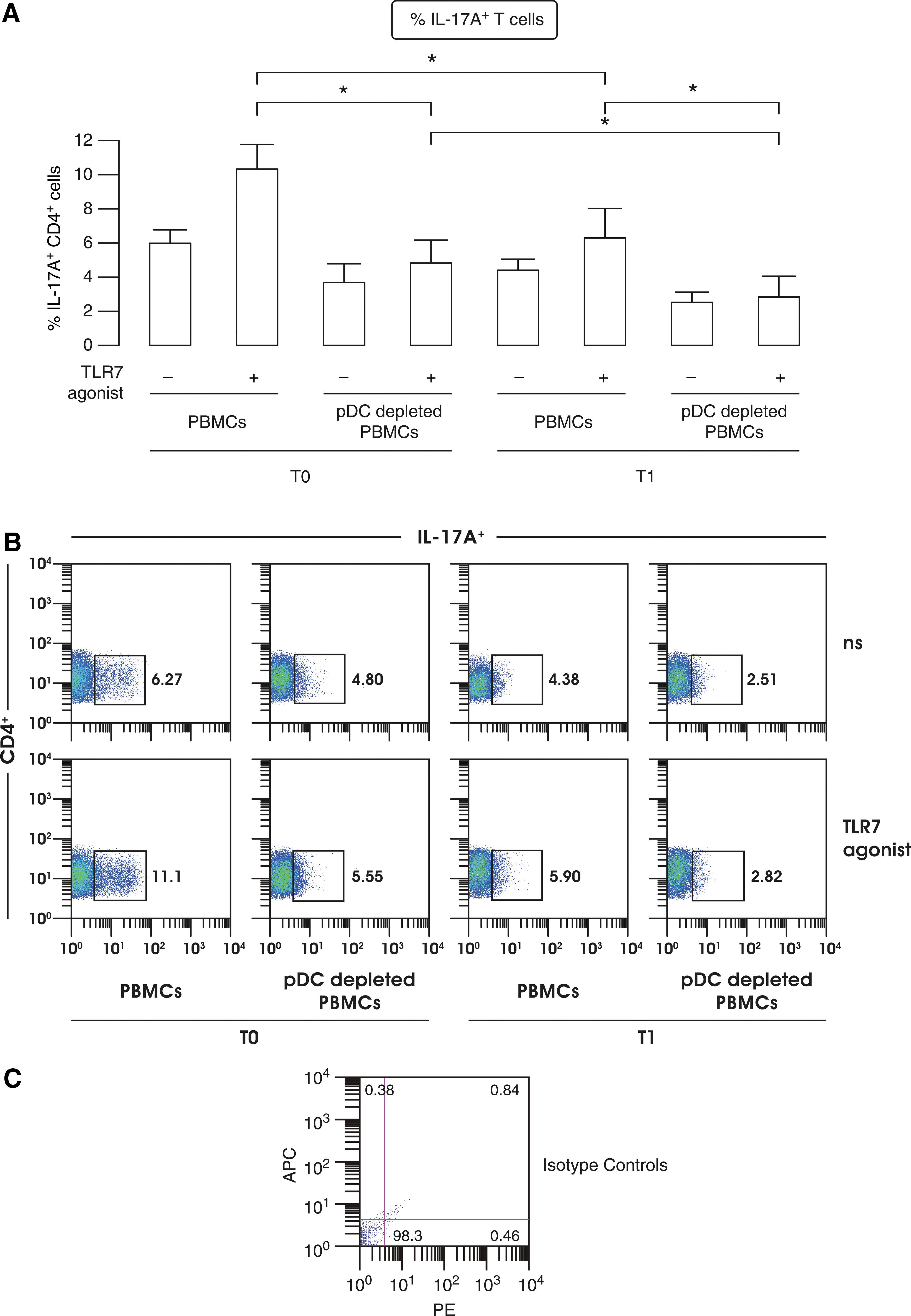
IFN-β therapy-mediated skewing of pDC-driven T-cell responses.
Furthermore, following in vivo IFN-β treatment the already decreased Th17 commitment was further reduced when pDCs were depleted. Another aspect to take into consideration is that in pDC-depleted PBMCs, both at T0 and T1, there was no induction of IL-17A+ T cells following TLR7 stimulation demonstrating that pDCs participate in the control of Th17 expansion.
The reduction of intracellular IL-17A expression in IFN-β-treated patients correlated with a reduced IL-17A release as measured by ELISA in supernatants of TLR7-treated PBMC cultures (Supplementary Fig. S3A). Accordingly, also an IFN-β-induced anti-inflammatory cytokine profile was found in this experimental setting, with reduced TNF-α release and concomitantly raised IL-10 levels upon TLR7 treatment (Supplementary Fig. S3B).
In line with the findings obtained in pDC-depleted PBMC cultures, the ability to commit Th17 polarization by MS-derived pDCs and the immuneregulation operated by IFN-β therapy on this cell type are even more clarified by the decrease in IL-17A production observed in mixed-leukocyte reaction consisting of in vivo IFN-β-exposed TLR7-treated pDCs cocultured with cord blood-derived allogeneic naïve CD4+ T cells (Supplementary Fig. S3C).
Thus, our findings suggest that IFN-β therapy might affect and modulate in vivo T-cell responses toward a more anti-inflammatory environment specifically regulating pDC immune functions.
Discussion
pDCs are immune cells with unique cell functions specialized in the secretion of type I IFNs following infection with most viruses. Although these features are of key importance for the induction of antiviral immune responses, chronic pDC activation and IFN production may result in the development of autoimmune diseases (Swiecki and Colonna 2010).
Recently, a pathogenic role was proposed for pDCs in the development of experimental autoimmune encephalomyelitis, a murine model of MS, by facilitating the induction of myelin oligodendrocyte glycoprotein-specific Th17 cells (Isaksson and others 2009). In the reported study, pDC depletion resulted in less severe clinical and histopathological signs of experimental autoimmune encephalomyelitis. Accordingly, pDC accumulation was found in brain lesions of MS patients (Serafini and others 2007; Lande and others 2008).
Different studies have characterized altered phenotype and functions in pDCs of MS patients when compared to healthy subjects. In particular, BDCA2+BDCA4+ pDCs were found in humans of at least 2 separate populations, CD123highCD58low pDC1 and CD123lowCD58high pDC2, mainly differing in the ability to induce IL-10-producing T reg or IL-17-producing Th17 cell differentiation, respectively (Schwab and others 2010). pDC1 and pDC2 were shown to have a reversed ratio in MS patients to that observed in healthy individuals with a higher frequency of pDC2, driving Th17 polarization. Furthermore, the ex vivo maturation status of these cells together with their ability to produce IFN-α and drive proliferative T-cell response and IFN-γ secretion in response to the TLR9 ligand CpG were found impaired in MS patients compared with controls (Stasiolek and others 2006; Sanna and others 2008; Bayas and others 2009; Hirotani and others 2012).
Our study characterized how IFN-β therapy, still one of the most important first-line treatments in use for RRMS, influences altered pDC-driven responses in MS patients and in particular those mediated by TLR7 triggering. This aspect was only partially characterized in patients versus controls (Mycko and others 2014) but it was never investigated before in detail in MS-affected individuals longitudinally studied before and after the beginning of IFN-β administration. Here, we highlighted a specific inhibition of TLR7-induced maturation and IFN-α production when pDCs from IFN-β-treated MS patients were stimulated in the context of PBMCs but not when these cells were isolated and cultured alone (compare Fig. 1 and Fig. 3A). These results demonstrate how a crosstalk with other immune cells via cell–cell contact or by bystander mechanisms might deeply influence outcome of pDC-mediated responses.
The best-known and studied IFN-β-induced anti-inflammatory cytokine is IL-10, which performs an irreplaceable role in negatively regulating inflammation (Murray 2006). Moreover, IL-10 signaling primarily functions to regulate DCs, in which IL-10 receptor expression is highest, and this role is mainly exerted by blocking the expression of specific subsets of TLR-induced genes at the level of transcription, including type I IFNs (Duramad and others 2003; Murray 2005).
In this study, we showed that in TLR7-treated PBMCs of MS patients the production of IFN-α, mediated by pDC stimulation, was dramatically reduced in the presence of IFN-β therapy (Fig. 3A) when the IL-10 release, mainly driven by monocytes, was strongest (Fig. 5A, B). As a further proof of the role of IL-10 in modulating pDC-mediated IFN-α expression upon IFN-β administration, the stimulation of TLR7 in isolated pDCs from MS patients showed an opposite trend in type I IFN release to that observed in PBMCs (compare Fig. 1 and Fig. 3A) and an in vitro treatment with IL-10 directly blocked IFN-α release by TLR7-treated pDCs (Fig. 5C).
The enhanced IFN-α induction in isolated in vivo IFN-β-exposed pDCs mirrored also the gene expression profile observed in these cells with an increased transcription of many factors involved in TLR stimulation and IFN production, such as TLR7 itself, MyD88, IRF7, and IRF5, and downregulation of the TLR signaling inhibitors Tollip and SOCS1 (Fig. 2). Results that matched recently published data on upregulation mediated by IFN-β therapy of TLR7 and MyD88 proteins in MS pDCs as evaluated by flow cytometry (Derkow and others 2013). Thus, here we report that IL-10 production, principally released by monocytes, was strikingly induced in PBMCs of IFN-β-treated patients upon TLR7 triggering and that this cytokine negatively regulates pDC-mediated IFN-α release. Taken together, these data demonstrate that pDCs of MS patients by themselves are actually more prone to produce type I IFN upon IFN-β therapy, conversely to what observed in the context of whole PBMCs.
Using the same setting, elsewhere we also demonstrated how IFN-β therapy is able to influence monocyte-B cell crosstalk in MS patients replenishing the impaired TLR7 responsiveness and TLR7-induced cytokine release, in turn, restoring a correct B-cell response and Ig production (Giacomini and others 2013). Hence, our experimental approach, consisting of mixed cell types in whole PBMC cultures, allowed the study of immune cells in a physiological setting more closely resembling what found in the periphery of patients and led to the dissection of immune-regulatory aspects, including the crosstalk of different cell types as a result of bystander responses regulated by IFN-β administration, which cannot be taken into account when studying purified cells.
Consistent with this view, IFN-β may correct and shape pDC-driven immune responses toward a more anti-inflammatory phenotype and, thus, IFN-β-modulated pDCs might also regulate T-cell differentiation upon TLR7 stimulation. Indeed, while the maturation of these cells is reduced in the presence of IFN-β, there is an induction of both ICOS-L and B7H1 expression (Fig. 6A, B), two members of the B7 family of immune-regulatory ligands that deliver co-inhibitory signals to T cells and that can contribute to the poor stimulatory capacity of immature pDCs (Collins and others 2005; Severa and others 2012). This IFN-β-induced phenotype of pDCs, and in particular the expression of ICOS-L, might also promote the generation of IL-10-producing T reg cells from naïve T cells, a subset of T cells necessary and sufficient to prevent autoimmunity throughout the lifespan of an individual (Ito and others 2007; Korn and others 2007). This hypothesis is in line also with the data from Schwab and others (2010) showing that in vivo IFN-β treatment can restore the imbalance in the pDC1/pDC2 ratio observed in MS patients, resulting in an increased T reg differentiation and in a suppressed Th17 commitment.
More interestingly, our data showing the presence of high level of IL-10 together with the raised pDC-mediated IL-27 expression and reduced IL-23 upon IFN-β therapy suggest the presence of an unfavorable milieu for Th17 cell differentiation in the context of TLR7 triggering, a possible therapeutic mechanism of IFN-β targeting pDC-triggered autoimmune response in MS. Indeed, Th17 cells are a distinct lineage of CD4+ T helper cells that have been implicated in the pathogenesis of different autoimmune diseases, including MS (Sallusto and Lanzavecchia 2009). In particular, IL-17A gene and protein expression is elevated in active MS brain lesions, and in mononuclear cells derived from blood or CSF of MS patients in comparison with controls (Matusevicius and others 1999; Lock and others 2002; Tzartos and others 2008; Montes and others 2009). Furthermore, studies performed in the MS mouse model, suggested that transfer of Th17 cells can induce tissue damage and that inflammation in the brain parenchyma occurs only when infiltrating Th17 cells outnumber Th1, thus driving a disproportionate increase in IL-17 expression in the brain (Luger and others 2008; Stromnes and others 2008). Th17 cell differentiation in humans is orchestrated by IL-1β and IL-23, which stimulate, and by IL-10 and IL-27, which inhibit the differentiation of this cell subset (Chen and O'Shea 2008; Volpe and others 2009; Segura and others 2013). Different reports have demonstrated that IFN-β can affect and inhibit at several levels the differentiation of Th17 cells both in the MS mouse model (Pennell and Fish 2014) and in MS patients. In particular, IFN-β therapy downregulated the expression of IL-1β and the p19 subunit of IL-23 in monocyte-derived DCs from MS-affected individuals and concomitantly induces the p28 subunit of IL-27 (Ramgolam and others 2009).
The IL-17A level present in sera or evaluated in myelin basic protein- or myelin oligodendrocyte glycoprotein-treated PBMCs, resulted higher in patients nonresponders to IFN-β therapy and lower in responder individuals (Balasa and others 2012; Kvarnstrom and others 2012). Furthermore, Zhang and others (2009) recently showed that IFN-β-induced TLR7 expression in monocyte-derived DCs is necessary for inhibition of IL-1β and IL-23 and induction of IL-27 secretion.
In line with these evidences, we found that pDCs derived from IFN-β-treated MS patients display an induced expression of both p28 and EBI3 IL-27 subunits and a reduction of IL23-p19 (Fig. 6C). The high production of IL-17A observed in TLR7-treated PBMCs from MS patients is reduced in the presence of IFN-β treatment, consistently with the promotion of an anti-inflammatory status resulting in a reduced TNF-α and an increased IL-10 release in the same cultures (Fig. 7 and Supplementary Fig. S3). More importantly, then, the depletion of pDCs from PBMCs specifically reduced the frequency of Th17 cells, demonstrating a key role of pDCs in the differentiation of Th17-skewed T cells and their likely pathogenic role in MS disease, as also recently suggested by Markovic-Plese's group that identified a key role for pDC-mediated endogenous IFN-β in the regulation of Th17 responses in MS patients (Tao and others 2014).
All together, these evidences highlight how stringently IFN-β therapy may tune pDC functions in MS patients acting on the regulation of bystander responses toward an anti-inflammatory-skewed cytokine milieu and led us to hypothesize that the modulation of TLR7 responses might be considered effective in driving an anti-inflammatory activity in the treatment of MS.
Footnotes
Acknowledgments
This work was supported by the Italian Multiple Sclerosis Foundation projects Nos. 2009/R/7 and 2013/R/9 (to E.M.C.). We thank Rosella Mechelli, Giovanni Ristori, Giulia Coarelli, and Danila Vittori, who took care of patients and helped with sampling; Eugenio Morassi (Division Service for Data Management, Documentation, Library, and Publishing Activities, Istituto Superiore di Sanità, Rome, Italy) for preparing drawings and Elisabetta Volpe (Neuroimmunology Unit, IRCCS Santa Lucia Foundation, Rome, Italy) for helpful discussion.
Author Disclosure Statement
M. Salvetti received lecture fees from Biogen-Dompé, research support from Bayer-Schering, Biogen-Dompé, Merck-Serono, Sanofi-Aventis. E.M.C. is the PI of a sponsored research agreement between Istituto Superiore di Sanità and Novartis Vaccines and Diagnostics s.r.l.