
Abstract
The inflammasome is a multimeric protein complex required for interleukin (IL)-1β production. Upon lipopolysaccharide (LPS) triggering of toll-like receptor (TLR)-4 and subsequent ATP signaling, the NOD-like receptor containing-pyrin domain 3 (NLRP3) inflammasome is activated to cleave pro-caspase-1 into caspase-1, allowing the secretion of IL-1β. IL-1β is known to function with IL-23 in the regulation of IL-17-producing CD4+ T cells, Th17 cells, in adaptive immunity. Recently, studies have shown that IL-1β and IL-23 together activate IL-17-producing innate lymphoid cells, demonstrating that the pair may exhibit additional effects on cell differentiation. Using an in vitro model of bacterial infection, LPS treatment of human monocytic cells, we investigated the molecular mechanisms involved in the co-expression of IL-1β and IL-23. We found that IL-1β is partially required for optimal LPS-induced IL-23 production. We also found that IL-23 production was partially dependent on ATP signaling via the P2X7 receptor, whereas IL-1β production required this signaling. Furthermore, we identified a novel role for cathepsin B activity in IL-23 production. Taken together, this study identifies differential requirements for the co-expression of IL-1β and IL-23. Due to their similar roles in Th17 differentiation, characterization of the regulatory mechanisms for LPS-induced IL-1β and IL-23 may reveal novel information into the pathology of the inflammatory response particularly during bacterial infection.
Introduction
I
In addition to caspase-1, the protease cathepsin B has also been identified as a potential requirement for both IL-1β and IL-18 production in response to a variety of PAMPs (Hentze and others 2003; Gicquel and others 2015a; Okinaga and others 2015). Although the mechanism remains unknown, cathepsin B has been proposed to exit the lysosome and act upstream of the NLRP3 inflammasome to regulate caspase-1 maturation and proinflammatory cytokine production (Hornung and others 2008). Furthermore, research has demonstrated a role for cathepsin B activity in the secretion of the prototypical proinflammatory cytokine tumor necrosis factor (TNF)-α (Ha and others 2008).
The IL-12 family of cytokines is composed of IL-12, IL-23, IL-27, and IL-35, structurally homologous, heterodimeric proteins composed of α- and β-subunits (Gee and others 2009). These cytokines are produced by professional antigen-presenting cells in response to pathogen and host-derived factors. Once produced, the IL-12 cytokines play an important role in the differentiation and maturation of CD4+ helper T cells. IL-23 is composed of a p19 subunit and the IL-12/23p40 subunit (Oppmann and others 2000). The hallmark function of IL-23 is defined by its ability to induce the differentiation of helper CD4+ T (Th) cells into the IL-17-producing Th17 cells (Hunter 2005; Kastelein and others 2007).
IL-1β functions in conjunction with IL-23 to regulate CD4+ T cell differentiation along the Th17 lineage (Coccia and others 2012a; Vignali and Kuchroo 2012). Furthermore, synergistic activity between IL-1β and IL-23 in the polarization of Th17 cells and IL-17 production has been reported (Sutton and others 2009a). A role for IL-1β in the regulation of IL-23 expression has been demonstrated in macrophages and dendritic cells (Peral de and others 2012a). This indicates that expression of these two cytokines may be linked via common regulatory mechanisms, one of which is the NLRP3 inflammasome. Herein, we demonstrate that IL-1β and IL-23 are co-expressed in human monocytic cells in response to LPS. We define a partial role for the NLRP3 inflammasome that works in conjunction with cathepsin B activation to modulate IL-23 expression.
Materials and Methods
Cell culture and reagents
The pro-monocytic leukemia cell line, THP-1, transfected with CD14-expressing cDNA plasmids (CD14 THP-1), was kindly provided by Dr. R Ulevitch (The Scripps Research Institute, La Jolla, CA). Parental THP-1 cells were purchased from American Type Culture Collection. Cells were cultured in Roswell Park Memorial Institute (RPMI) medium (Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and incubated at 37°C with 5% CO2. Cells were cultured at 1 × 106 cells/mL and stimulated with 1 μg/mL 0.111:B4 Escherichia coli LPS (Sigma-Aldrich, St. Louis, MO). Cells were pretreated with various antagonists over a range of doses: the caspase-1 inhibitor, Z-WEHD-FMK (R&D Systems, Minneapolis, MN), the NLRP3 inhibitor, glybenclamide (InvivoGen, San Diego, CA), the cathepsin B inhibitor, CA-074 (Tocris Bioscience, Bristol, United Kingdom), IL-1β neutralizing antibodies (R&D Systems) or the ATP receptor antagonists KN-62 or AZ 11645373 (Tocris Bioscience). Recombinant IL-1β was purchased from R&D Systems. Unless otherwise indicated, all medium control wells were treated with equivalent amounts of DMSO or PBS as appropriate controls for chemical inhibitors.
Monocyte isolation
Whole blood from healthy donors was obtained under Queen's University Research Ethics Board approval in heparin-coated tubes. Whole blood samples were enriched with the RosetteSep™ Human Monocyte Enrichment Cocktail (StemCell Technologies, Vancouver, BC) as described by the manufacturer's instructions. Enriched blood was overlaid on Lympholyte Human Cell Separation Media (Cedarlane, Burlington, ON) and processed by density centrifugation for 20 min at 800 g. Cells were washed twice with PBS plus 10 mM EDTA and 2% FBS, then resuspended in RPMI +10% FBS at 1 × 106 cells/mL.
Cytokine enzyme-linked immunosorbent assays
CD14 THP-1 cell supernatants were used for enzyme-linked immunosorbent assays (ELISA) for IL-1β, IL-12/23p40, and IL-23. ELISA protocols and reagents were obtained from eBioscience, San Diego, CA (IL-1β: 88-7010-88, IL-23: 88-7237-88, IL-12/23p40:BMS2013MST). All assays were performed as per manufacturer's instructions. Absorbancies were read at 450nm on a BioTek EL800 (BioTek Instruments, Winooski, VT). Data are presented as the mean ± standard deviation of technical replicates and are representative of at least 3 independent experiments.
Flow cytometry
Cells were resuspended in 200 μL 1× PBS-azide (Bioshop Canada, Inc., Burlington, ON) containing 2% FBS and stained with LPS from E. coli Serotype 055:B5, Alexa Fluor® 488 Conjugate (Molecular Probes, Eugene, OR) for 10 min at 37°C or anti-human TLR-4 Alexa Fluor 488 (eBioscience) and anti-human CD14 Biotin (eBioscience) for 30 min on ice or anti-human P2X7 (Developmental Studies Hybridoma Bank [DSHB]) for 10 min at room temperature. P2X7-L4 was deposited to the DSHB by Gu, B. (DSHB Hybridoma Product P2X7-L4). Cells were also stained with streptavidin Alexa Fluor 610-R-phycoerythrin conjugate (Molecular Probes) or R-phycoerythrin goat anti-mouse IgG (H+L) (Biotium, Hayward, CA) secondary antibody for 30 min on ice. Data were acquired with Coulter epics XL (Beckmann Coulter, Miami, FL). Data were analyzed with FlowJo ×10.0.7r2 software.
ATP ASSAY
ATP secretion was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI). Briefly, cell-free supernatants were added to an opaque 96-well plate and incubated at room temperature for 30 min. An equal volume of CellTiter-Glo reagent was added to each well and plates were incubated for 10 min at room temperature. Luminescence was recorded using a GloMax 96 Microplate Luminometer (Promega) with an integration time of 1 s per well. Data are presented as the fold increase over background luminescence for ATP secretion from at least 10 separate stimulations with data collected in duplicate. Error bars represent the standard error of the mean across all data sets.
Statistical analysis
Statistical analysis was calculated using a 2-tailed unpaired student's t-test assuming unequal variance. Statistical analysis is denoted on each figure: *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant.
Results
LPS induces IL-1β and IL-23 expression in CD14 transfected THP-1 cells
As an in vitro model of primary human monocytes we used THP-1 cells stably transfected with CD14 (CD14 THP-1) (Ma and others 2001). To confirm CD14 expression levels in these cells, parental THP-1 cells, CD14 THP-1 cells and primary human monocytes were stained for CD14 and TLR4 surface expression. Total TLR4 expression (quadrant [Q] 2 and Q3) was found to be higher in the parental THP-1 cells than CD14 THP-1 cells (65.7% versus 12.96%), which is more similar to the levels observed in primary monocytes (0.86%) (Fig 1A, left panels). CD14 THP-1 cells were 79.9% positive for total CD14 expression (Q1 and Q2), similar to primary monocytes that were 76.8% positive for CD14, whereas parental THP-1 cells were only 15.8% positive for CD14 (Fig. 1A, right panels). As a consequence, the CD14 THP-1 cells demonstrated higher LPS binding compared with the parental THP-1 cells (Fig. 1B, C). Therefore, to study the mechanisms regulating LPS-induced IL-23 and IL-1β expression in human monocytes, we used CD14 THP-1 cells as our model system.
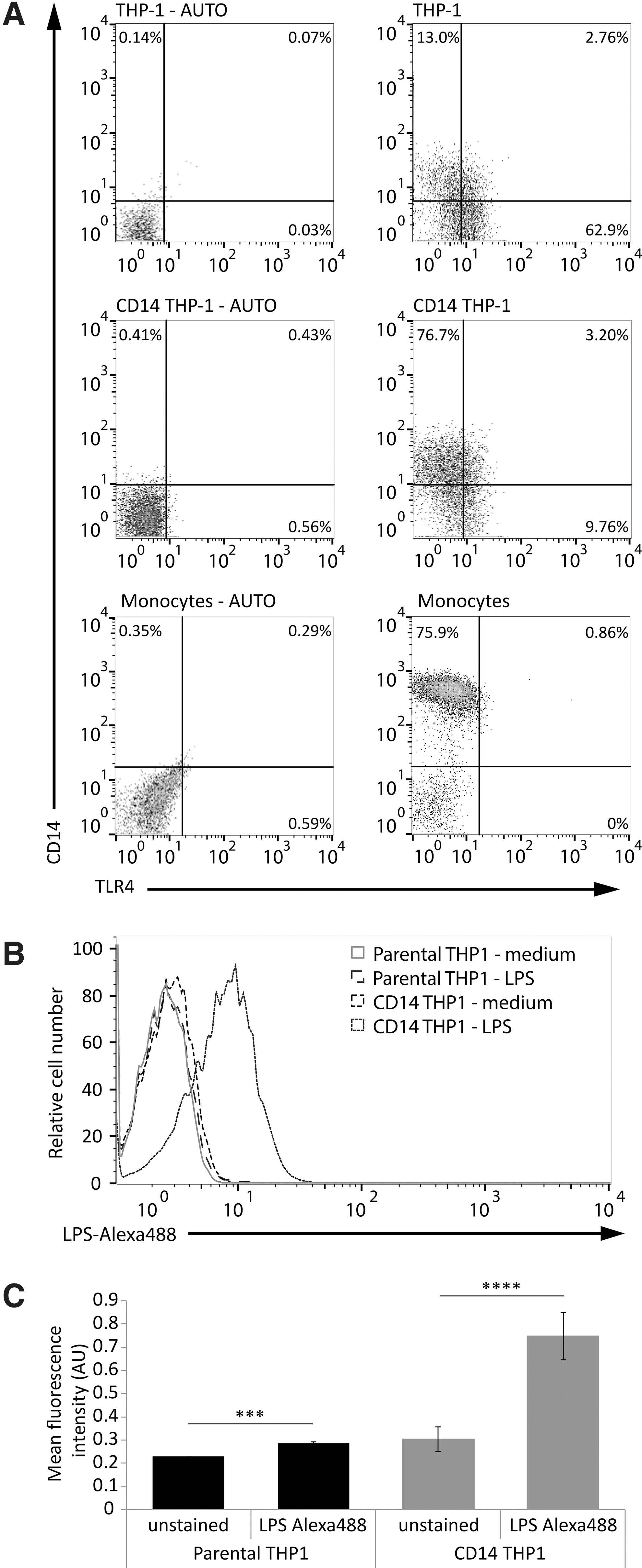
CD14 THP-1 cells express higher levels of surface CD14 and bind higher levels of lipopolysaccharide (LPS) compared with parental THP-1 cells. Cells were harvested and stained for CD14 and TLR4 expression before analysis by flow cytometry
Initially, we performed time course experiments (0, 4, 8, and 16 h) with and without LPS treatment to identify the optimal time point for cytokine production. Since the IL-12/23p40 subunit is also induced upon LPS stimulation in human monocytes (Ma and others 2004) and its expression is required for bioactive IL-23, we measured expression of IL-12/23p40 in addition to that of IL-23 and IL-1β. Since IL-12p70 and IL-23 share the IL-12/23p40 subunit, we measured secretion of IL-12p70, which remained undetectable at all time points (data not shown). IL-23 expression was detected as early as 4 h post-LPS treatment (Fig. 2A); as was IL-12/23p40 expression (Fig. 2B). Release of IL-1β was detected at 16 h post-LPS stimulation (Fig. 2C). Since all cytokines exhibited significantly high expression at 16 h, we chose this time point to further investigate co-regulation of IL-23 and IL-1β in response to LPS stimulation.
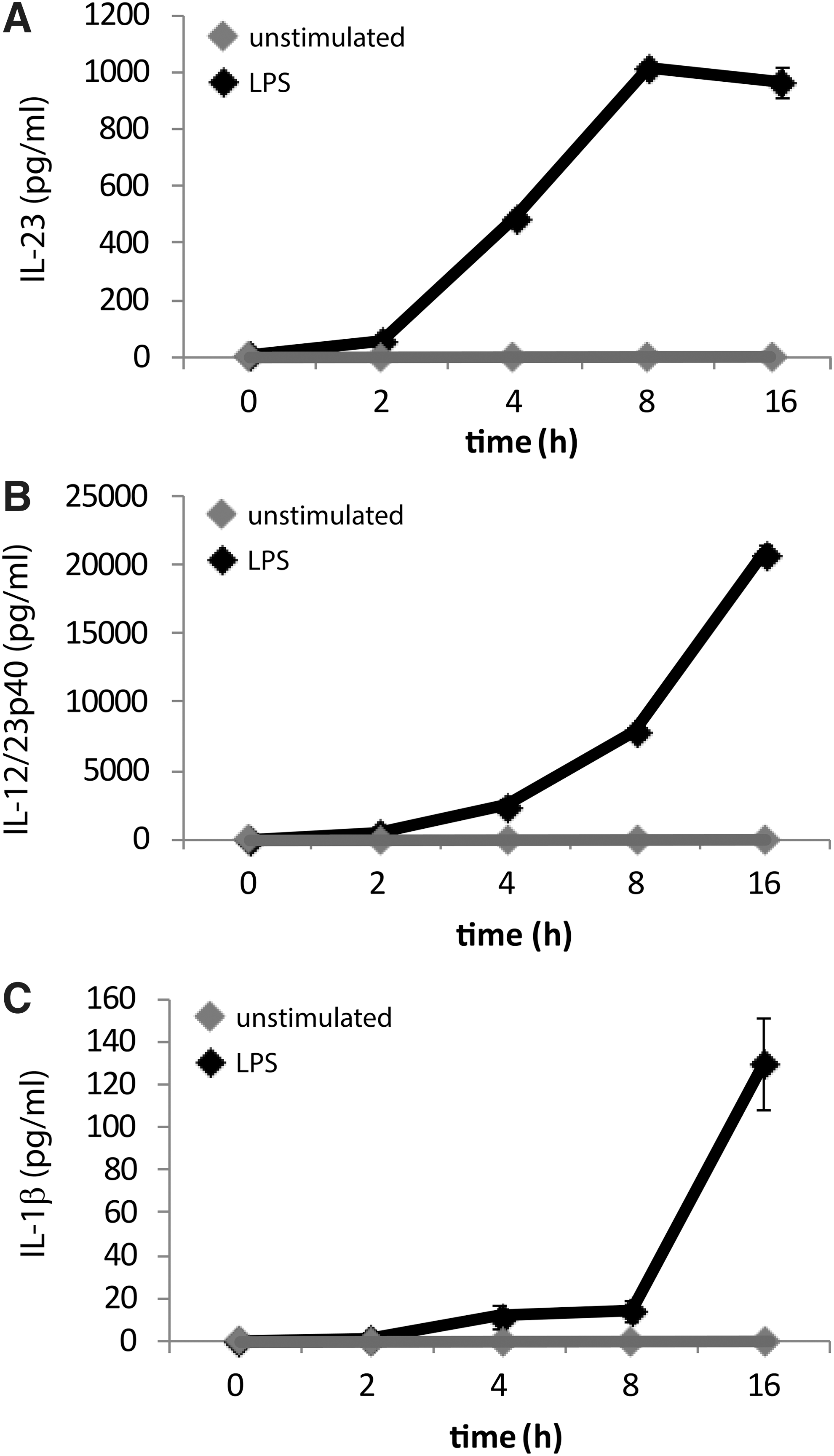
LPS induces interleukin (IL)-23, IL-12p40, and IL-1β concurrently in CD14 THP-1 cells. Cells were treated for times ranging from 0 to 16 h with or without LPS (1 μg/mL). IL-23
IL-23 expression is partially dependent on IL-1β secretion and NLRP3 activation
Previous studies have demonstrated that IL-1β production is required, at least in part, for IL-23 secretion in PMA-matured THP-1 cells (Peral de and others 2012a), therefore we tested if this was the case in both untreated and PMA-treated CD14 THP-1 cells. CD14 THP-1 cells were cultured in PMA (10 ng/mL) for 48 h and then treated with LPS for 16 h in the presence of a neutralizing antibody for IL-1β and resulting IL-23 and IL-12/23p40 secretion was measured by ELISA. The data show that IL-23 and IL-12/23p40 are inhibited significantly at the highest concentration of anti-IL-1β antibody used in PMA-treated CD14 THP-1 cells (Fig. 3A). In untreated CD14 THP-1 cells, IL-23 and IL-12p40 production is reduced with the addition of 100 ng/mL of the IL-1β neutralizing antibody, however, only the reduction in IL-23 production reached statistical significance (Fig. 3B). Interestingly, in both cell types this inhibition is not complete and thus indicates that IL-23 production is only partially dependent on IL-1β secretion.
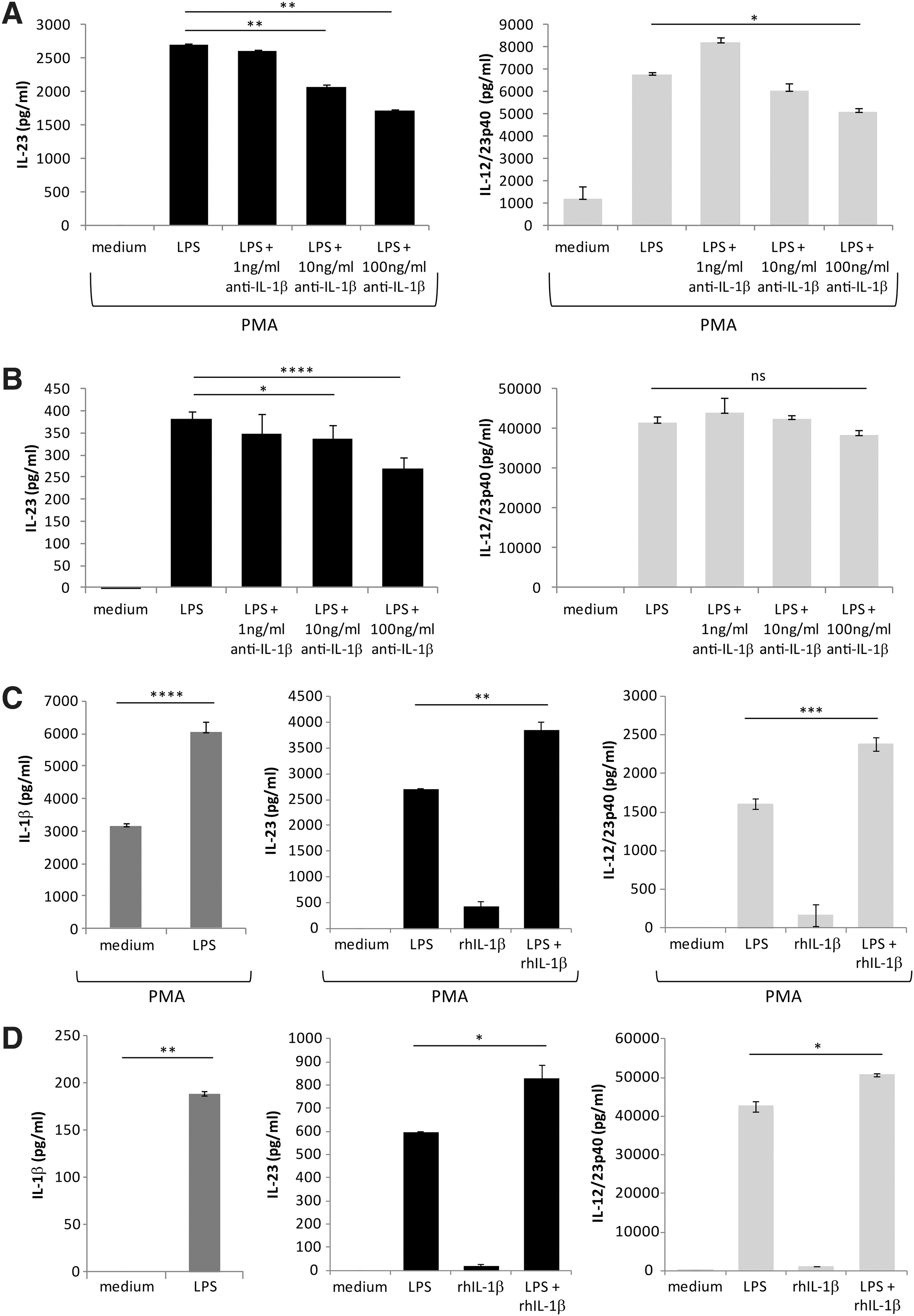
LPS-induced IL-23 and IL-12p40 is partially dependent on endogenously produced IL-1β. Cells were either pretreated with PMA (10 ng/mL) for 2 days
In comparison to the PMA-treated cells, the undifferentiated CD14 THP-1 cells exhibited less of a decrease in IL-23 and IL-12/23p40 expression in the presence of the IL-1β neutralizing antibody. We reasoned that this may be due to differential amounts of IL-1β release between the two cell types. Thus, we measured IL-1β secretion by ELISA on LPS treated PMA-differentiated and undifferentiated CD14 THP-1 cells (Fig. 3C, D; left panels). Under these conditions, we observed that the PMA-treated CD14 THP-1 cells produced very high amounts of IL-1β even in the absence of LPS, whereas undifferentiated CD14 THP-1 cells did not. Upon stimulation with LPS, PMA-treated cells exhibited a 2-fold increase in IL-1β production compared to unstimulated controls. To confirm that IL-1β induces upregulation of IL-23 and IL-12/23p40, we treated CD14 THP-1 cells or PMA-treated CD14 THP-1 cells with recombinant IL-1β in the presence and absence of LPS (Fig. 3C, D; middle and right panels). The data show that while IL-1β alone is insufficient to induce either IL-23 or IL-12/23p40 expression in either cell type, when given IL-1β with LPS, both IL-23 and IL-12/23p40 are significantly upregulated relative to LPS alone. Taken together, these results suggest that in response to LPS, IL-1β functions to promote IL-23 and IL-12/23p40 expression.
To explore whether inflammasome induction is involved in IL-23 production, we treated CD14 THP-1 cells with LPS in the presence of the NLRP3 inflammasome inhibitor glybenclamide (Lamkanfi and others 2009a). Our data show that IL-23 induction by LPS is partially inhibited at low doses of glybenclamide (1 and 10 μg/mL) and is enhanced at higher doses (25 μg/mL) (Fig. 4A). Interestingly, when we examined IL-12/23p40 expression under these conditions, there was no significant impact of glybenclamide on IL-12/23p40 expression (Fig. 4B). To confirm that glybenclamide was indeed inhibiting inflammasome activation, IL-1β secretion was measured (Fig. 4C). As expected, IL-1β was significantly downregulated by glybenclamide in a dose-dependent manner. Taken together, these data indicate that IL-12/23p40 induction is not directly affected by NLRP3 blockade, whereas formation and release of IL-23 may partially depend on NLRP3 inflammasome activation.
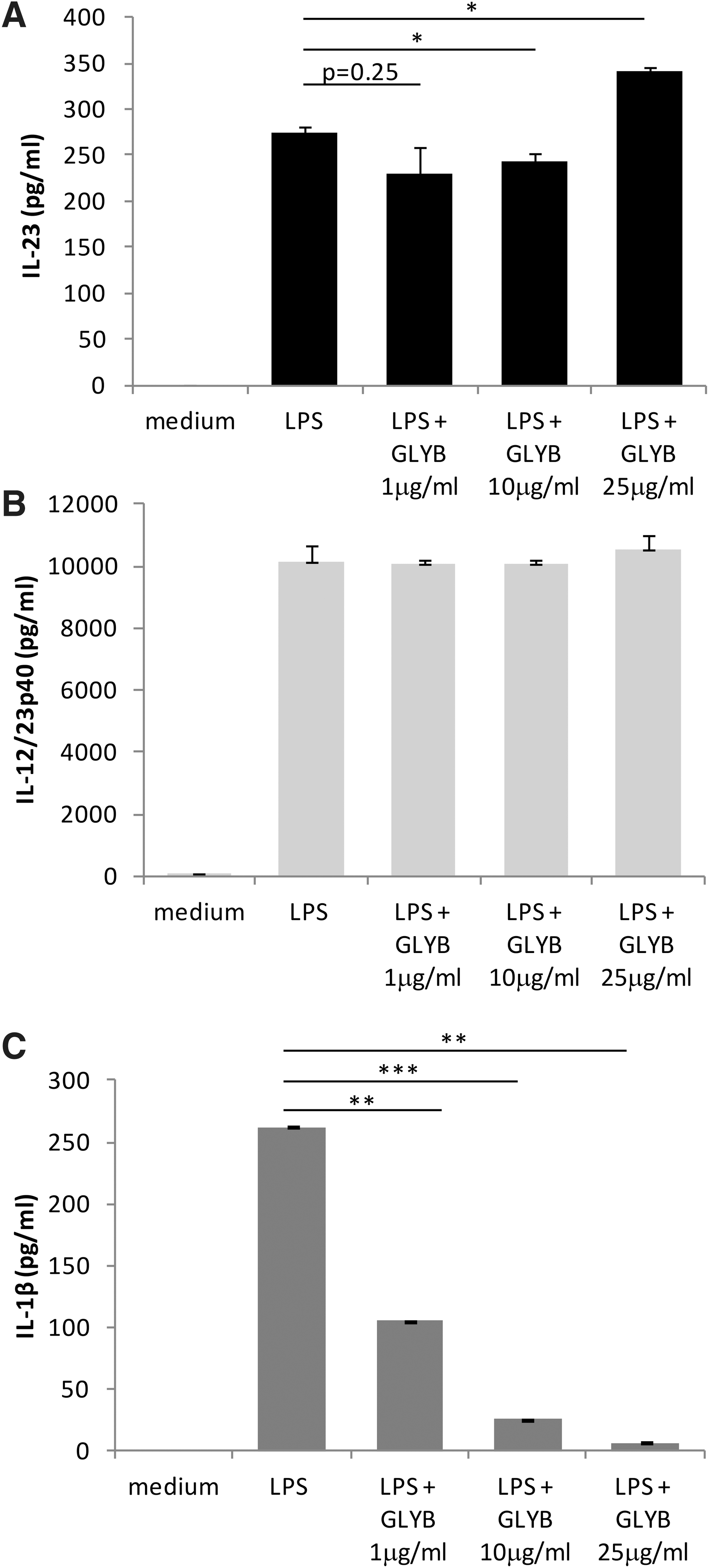
LPS-induced IL-23 and IL-12p40 is not dependent on NLRP3 activity. Cells were treated for 16 h with LPS (1 μg/mL) in the presence of various doses of the NLRP3 inhibitor: glybenclamide (GLYB). IL-23
LPS enhances ATP release in CD14 THP-1 cells but ATP-signaling is only partially involved in IL-23 induction
Since ATP has been previously shown to enhance IL-23 expression in dendritic cells (Schnurr and others 2005), and ATP signaling provides signal 2 for inflammasome induction (Perregaux and others 2000), we examined whether LPS stimulation was able to induce ATP release from CD14 THP-1 cells. In cells treated with LPS for 16 h, we observed a significant enhancement of ATP release (Fig. 5A). Since LPS enhanced the amount of ATP released from the cells, we also examined whether LPS could modulate the expression of the ATP receptor, P2X7. In cells treated with LPS for 16 h, a significant increase in P2X7 expression was observed, from 11.1% in untreated cells to 19.3% with LPS stimulation (Fig. 5B). To examine whether endogenous ATP signaling is involved in LPS-mediated induction of IL-23 expression, we treated cells with either KN-62 (KN), a P2X inhibitor that functions to block CaM kinase II activity (Gargett and Wiley 1997), or AZ11645373 (AZ), a selective P2X7 antagonist (Stokes and others 2006), for 15 min before incubation with LPS for 16 h. IL-23 and IL-12/23p40 expression were significantly inhibited, although partially, by AZ but not KN (Fig. 5C). IL-23, but not IL-12/23p40, was also partially decreased in the presence of KN, although this decrease did not reach statistical significance. As a positive control, LPS-induced IL-1β expression was significantly decreased in the presence of either KN or AZ. Our data suggest that although LPS induces ATP release, this pathway may only play a partial role in the induction of IL-23 expression.
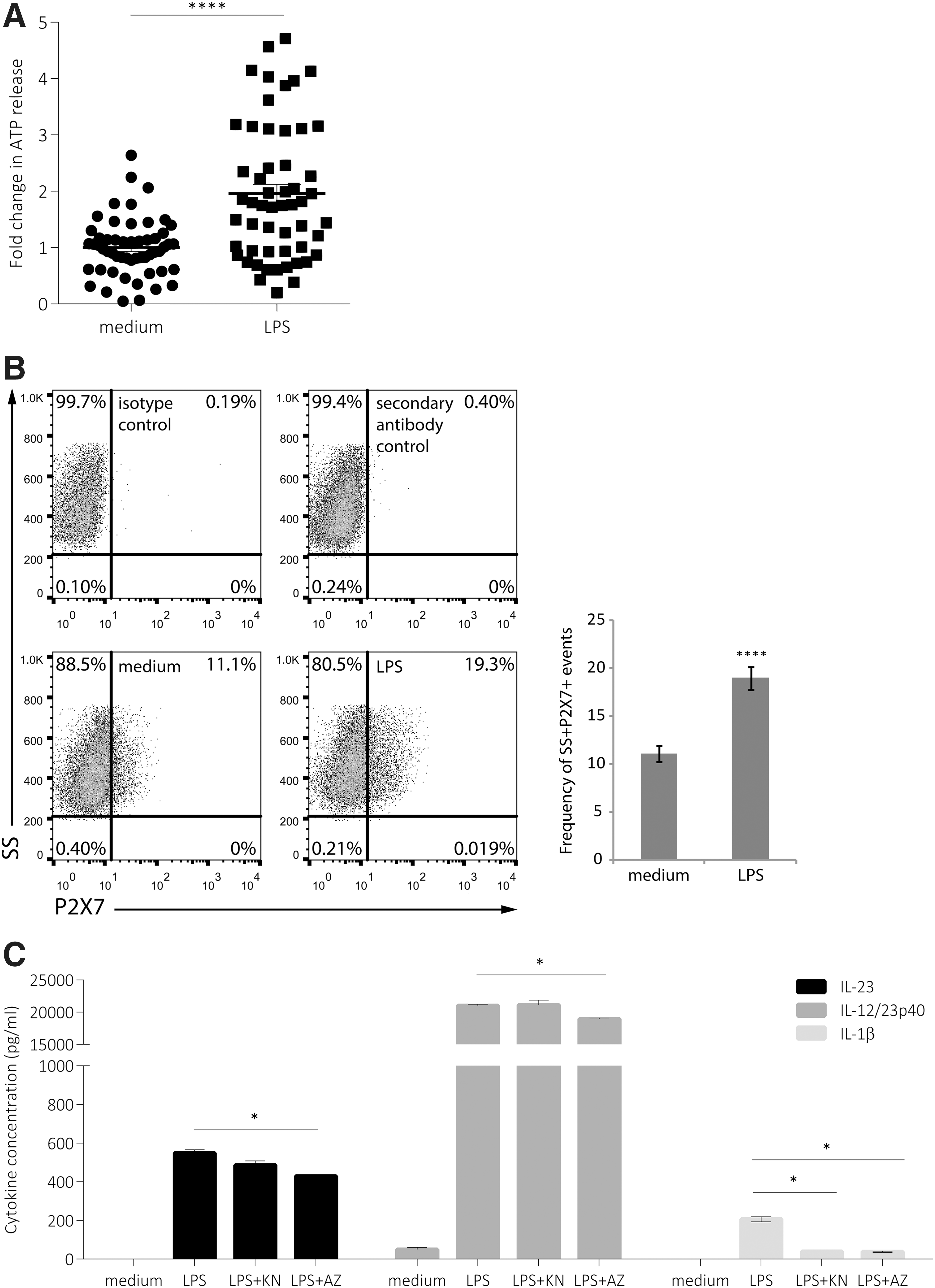
LPS-induced IL-23 and IL-12/23p40 is not dependent on P2X7 activity. Cells were treated for 16 h with LPS (1 μg/mL)
IL-23 expression is dependent on caspase-1 and cathepsin B activity
So far our data demonstrate a partial dependence of inflammasome and IL-1β induction for optimal IL-23 production. Next, we tested to see whether caspase-1, the enzyme necessary for cleavage and ultimate release of IL-1β, is required for IL-23 secretion. We examined CD14 THP-1 cells incubated in the presence of the caspase-1 inhibitor Z-WEHD-FMK (FMK) for 15 min before LPS stimulation for 16 h. The results show that IL-23, IL-12/23p40, and IL-1β are sensitive to caspase-1 inhibition at 10 and 100 μM FMK (Fig. 6 A–C, left panels). The partial inhibition observed at 10 μM FMK indicates that IL-23 secretion may be at least partially dependent on caspase-1. However, inhibition of both IL-23 and IL-12/23p40 with 100 μM FMK was far greater than that observed with either the neutralizing antibody to IL-1β or KN/AZ inhibition. At high concentrations, FMK may have nonspecific effects on cathepsin activation; previous studies have indicated that due to the similarity in structure, caspase-1 inhibitors may also efficiently inhibit cathepsin B activity (Schotte and others 1999). To test whether IL-23 expression is sensitive to cathepsin B protease activity we treated cells with the cathepsin B inhibitor CA-074 (5 nM). We observed a striking decrease in expression of IL-23 but not IL-12/23p40 or IL-1β in the presence of CA-074. The data indicate that optimal IL-23 secretion is dependent on cathepsin B activation while IL-12/23p40 and IL-1β are not (Fig. 6A-C, right panels). Taken together these data indicate that secretion of bioactive IL-23 shares a partial requirement on inflammasome activation and IL-1β; however, IL-23 release is dependent on cathepsin B activity.
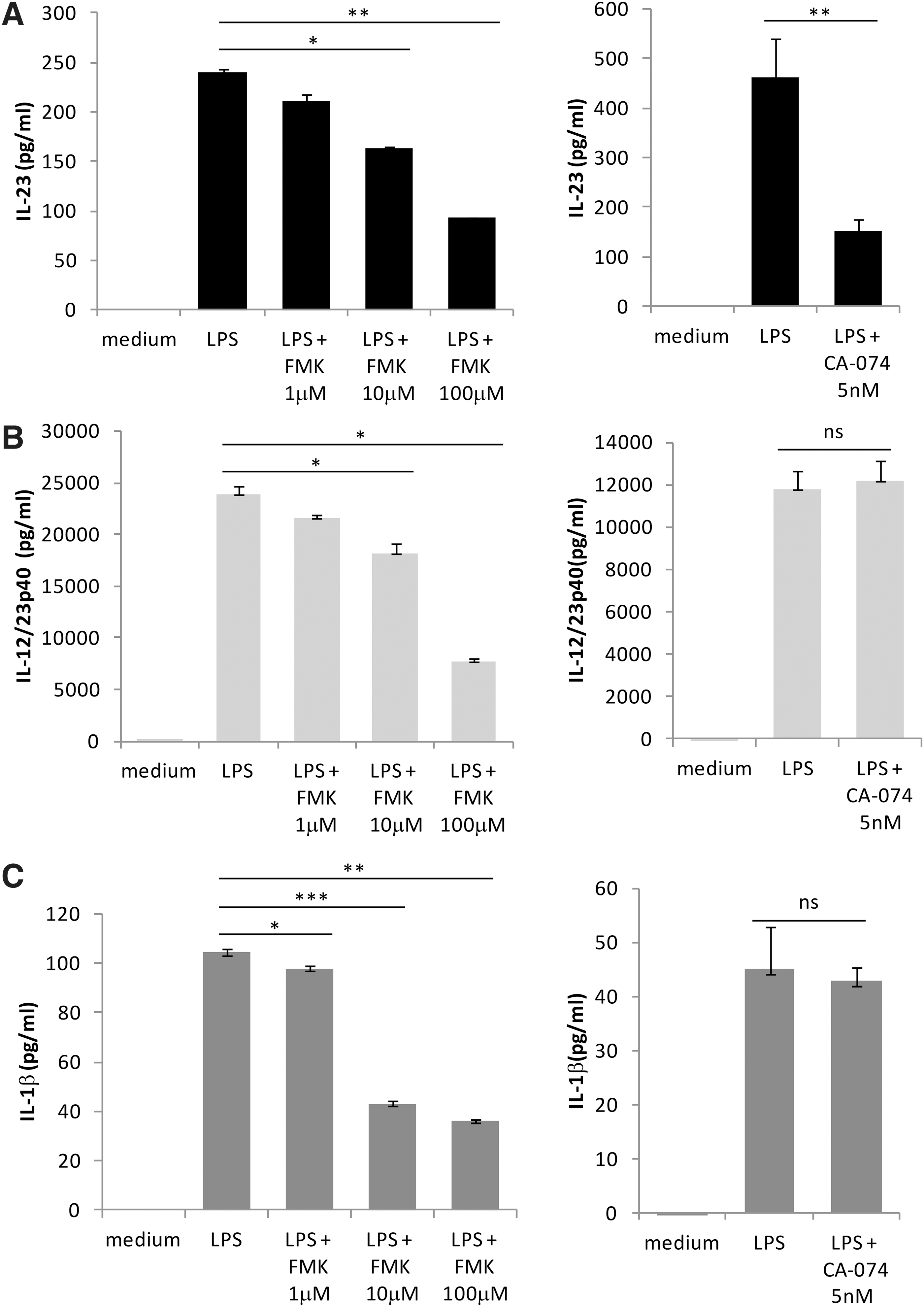
LPS-induced IL-23 expression is dependent on caspase-1 and cathepsin B activity. Cells were treated for 16 h with LPS (1 μg/mL) in the presence of the caspase inhibitor: Z-WEHD-FMK (FMK) (left panels) or the cathepsin B inhibitor CA-074 (right panels). IL-23
Discussion
In this study, we have identified that IL-23 expression can be regulated by multiple factors including inflammasome activation, ATP signaling, and, most strikingly, by cathepsin B activation. Understanding the mechanisms that control IL-23 and IL-1β expression levels is important in the context of regulation of inflammatory processes and inflammatory disease.
The dependence of IL-23 expression on IL-1β has not been fully elucidated to date. Our data demonstrating that IL-23 is at least partially dependent on autocrine-induced IL-1β in human monocytic cells parallels a similar human macrophage study (Peral de and others 2012a). In in vitro models, inhibition of autophagy is linked with enhancement of inflammasome activation and resulting IL-1β production (Harris and others 2011). In such a model, Peral de and others 2012b demonstrated a dependence of IL-23 expression on IL-1β. IL-1β induction has also been linked to IL-23 production in in human periodontal ligament fibroblasts (Zhu and others 2012).
Extracellular ATP is an NLRP3 agonist; when released from damaged cells, it can act as an autocrine or paracrine molecule, and it binds to the P2X7 ATP-gated ion channel, resulting in K+ efflux (Perregaux and Gabel 1994). Using glybenclamide, which blocks the NLRP3-specific potassium efflux channel (Lamkanfi and others 2009b), we demonstrated that IL-23 induction was significantly inhibited at relatively low concentrations of glybenclamide (10 μg/mL), but at higher concentrations (25 μg/mL), IL-23 expression was enhanced. Interestingly, IL-23 expression induced by Clostridium difficile toxins in murine dendritic cells was recently shown to be sensitive to glybenclamide treatment at 25 μg/mL (Cowardin and others 2015). It is possible that the increased levels of IL-23 expression observed at 25 μg/mL glybenclamide in our model could be due to the induction of alternative pathways, which may potentially be enhanced in THP-1 cells under suppression of K+ efflux.
A role for ATP-mediated signaling through the P2 receptors, as demonstrated by inhibition with the P2 antagonist pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS), along with forskolin and prostaglandin E2-mediated cAMP signaling, have been defined to play a role in induction of IL-23 (Schnurr and others 2005). Others have shown ATP-independent induction of calcium signaling pathways enhances IL-23 expression (Paustian and others 2013). Although LPS induced upregulation of P2X7 surface expression, we demonstrated a minor role for the P2X7 receptor in IL-23 and IL-12p40 induction through the use of the P2X7-specific inhibitors: KN and AZ. Since the KN inhibitor also blocks Ca2+ signaling by inhibiting CaM-kinase II activation, it is possible that induction of Ca2+ signaling may modestly effect IL-23 expression in our model. However, our focus was on studying the role of the P2X7 receptor on the co-expression of IL-1β and IL-23; we chose to use concentrations for which P2X7 was targeted (IC50 = 15 nM) (Chessell and others 1998; Stokes and others 2006). For inhibition of CaM kinase II, the IC50 = 0.9 mM (Tokumitsu and others 1990), therefore, we likely did not sufficiently inhibit Ca2+ signaling that may have resulted in a more dramatic effect of the inhibitor on IL-23 expression. Other pathways, such as the induction of MAPK activation have been shown to be required for TLR-mediated induction of IL-23 expression (Dobreva and others 2008; Utsugi and others 2006; Wang and others 2011). It is possible that cross-talk between calcium signaling and MAPK activation is responsible for controlling IL-23 expression downstream independently of ATP signaling and inflammasome activation.
Given that the regulation of IL-23 can be modulated at several levels, it is important to understand which of these mechanisms are critical to optimal IL-23 production. Our data demonstrate that unlike IL-23, production of IL-12/23p40 is independent of ATP-induced K+ efflux and IL-1β production in undifferentiated CD14 THP-1 cells. Interestingly, in PMA-treated CD14 THP-1 cells, both IL-23 and IL-12/23p40 were dependent on IL-1β production, possibly due to the higher level of IL-1β expression in these cells. We also demonstrated that release of IL-12/23p40 was independent of cathepsin B activity in CD14 THP-1 cells. Under conditions where caspase-1 activity was blocked via incubation with FMK, IL-23, IL-12/23p40, and IL-1β were inhibited, indicating that caspase-1 activity may be required for cytokine release from the cells. Indeed others have demonstrated inhibition of caspase-1 activity and IL-1β release from whole blood samples, macrophages, and THP-1 cells at 10–100 μM of FMK, as used in our study (Zhu and others 1995; Lipinska and others 2014; Gicquel and others 2015b).
We demonstrated that IL-23, but not IL-1β, was dependent on cathepsin B activity. Some reports have indicated that inflammasome activation can result from crystalline-induced disruption of lysosomal membranes resulting in the release of cathepsin B, which is postulated to be sensed by NLRP3 (Hornung and others 2008; Duewell and others 2010; Kuroda and others 2011; Yang and others 2014). In our system, we observed IL-1β production in the presence of CA-074, a cathepsin B inhibitor. In this study, we used comparably low doses of CA-074, (5 nM [1.9 ng/mL] versus 10–20 μg/mL methylated CA-074 (Kuroda and others 2011) to minimize nonspecific effects of the inhibitor. Furthermore, we used the nonmethylated form of CA-074 (IC50 = 2.24 nM) demonstrated to be specific for cathepsin B, while not affecting cathepsin L (Murata and others 1991; Montaser and others 2002). Therefore, differences between cell types and stimuli may account for the observed variability for the requirement of cathepsin B activity for IL-1β production. Other inflammasome agonists such as Aggregatibacter actinomycetemcomitans resulted in reactive oxygen species-dependent and cathepsin B-dependent IL-1β production without the requirement of caspase-1 activation (Okinaga and others 2015). However, the precise mechanism of cathepsin B recognition by NLRP3 remains unclear, and in agreement with our data, several studies have shown that inflammasome activation can occur independently of cathepsin B activation (Dostert and others 2009; Newman and others 2009; Munoz-Planillo and others 2013; Hari and others 2014).
The mechanism behind cathepsin B-mediated IL-23 expression as observed in our study remains unknown. Cathepsin B activity has been implicated in LPS-mediated TNF-α release, where Ha and others 2008 demonstrated a role for cathepsin B in post-translational modifications and trafficking of TNF-α for secretion. Since we showed a differential requirement for cathepsin B activity between IL-23 and IL-12/23p40, this indicates a potential role for cathepsin B in regulating release of preformed IL-23 (pre-association of IL-23p19 and IL-12/23p40). Similarly, dimerization of IL-12p35 and IL-12/23p40 occurs in the endoplasmic reticulum (Carra and others 2000) and is followed by release of bound subunits as IL-12p70 by the “unit-interaction” secretion pathway (Duitman and others 2011). Indeed, the p40 subunit has been shown to stabilize IL-12p35 for release of IL-12p70 (Jalah and others 2013). However, the precise mechanisms governing the regulation of how the subunits combine to form either IL-23 or IL-12p70 have yet to be determined.
The dependence, although partial, of IL-23 on IL-1β expression and differential requirements for the ATP/P2X7 and cathepsin B activity that we observed could have vast ramifications on how the adaptive arm of the immune system responds to infection or in patients with autoimmune disease. A particular influence on the formation of Th17 cells has been widely described in the literature for IL-23. More recently, IL-1β in tandem with IL-23 has been demonstrated to play a significant role in the induction and differentiation of Th17 cells and, in particular, the development of pathogenic Th1/Th17 cells (Barrie and others 2011; Shainheit and others 2011; Zielinski and others 2012; Duhen and Campbell 2014).
As well, NKT cell, invariant NKT cell, and gamma/delta (γδ) T cell-mediated IL-17 production require both IL-1β and IL-23 (Sutton and others 2009b; Doisne and others 2011; Moreira-Teixeira and others 2011). A novel role for both IL-23 and IL-1β in innate lymphoid cell (ILC) development has been recently described (Coccia and others 2012b). ILC3 cells are known to respond to IL-1β and IL-23 (Ward and Umetsu 2014) and stimulation of ILC1 with IL-2, IL-1β, and IL-23 differentiated the cells to shift to an ILC3 phenotype (Bernink and others 2015). Taken together, this study provides novel information on the complex interplay between the regulation of IL-1β and IL-23 expression, which is critical to identifying potential therapeutic targets for inflammatory disease.
Footnotes
Acknowledgments
This research was funded by the National Sciences and Engineering Research Council of Canada (NSERC). C.W. was supported by an NSERC Alexander Graham Bell studentship. C.P. was supported by an Ontario Graduate Scholarship and the Dr. Robert John Wilson Graduate Fellowship. A.T. was supported by an NSERC undergraduate student research award.
Author Disclosure Statement
No competing financial interests exist.