
Abstract
The study objective was to compare the efficacy and safety of peginterferon lambda-1a combined with ribavirin/daclatasvir (Lambda/RBV/DCV), versus peginterferon alfa-2a combined with ribavirin/telaprevir (Alfa/RBV/TVR), in patients chronically infected with hepatitis C virus (HCV), genotype 1b. This was a prospective, randomized, open-label, phase 3 study (NCT01718158) in adults (aged ≥18 years) who were treatment naïve or prior relapsers to peginterferon alfa/ribavirin therapy. The primary endpoint was sustained virologic response at post-treatment follow-up week 12 (SVR12). Patients were randomized in a 2:1 ratio to receive 24 weeks of Lambda/RBV/DCV or response-guided 24 or 48 weeks of Alfa/RBV/TVR. Overall, 440 patients were treated (294 with Lambda/RBV/DCV; 146 with Alfa/RBV/TVR). The proportion of patients achieving SVR12 was 88.8% in the Lambda/RBV/DCV arm and 70.5% in the Alfa/RBV/TVR arm (difference between arms: 18.3%; 95% confidence interval: 9.9–25.7; P < 0.0001). Patients in the Lambda/RBV/DCV group had fewer rash-related adverse events (AEs), cytopenic abnormalities, flu-like symptoms, serious AEs, and discontinuations due to AEs, but more liver abnormalities than those in the Alfa/RBV/TVR group. In conclusion, treatment with Lambda/RBV/DCV led to higher SVR12 rates and a more favorable safety profile than Alfa/RBV/TVR in patients with chronic HCV, genotype 1b infection.
Introduction
C
A potential alternative, type III lambda interferons, were identified in 2003 (Kotenko and others 2003; Sheppard and others 2003) and found to have similar biologic and intracellular antiviral activities to the type I alfa interferons (Kotenko and others 2003; Dumoutier and others 2004; Zhou and others 2007). The 2 classes of interferons differ, however, with regard to receptor distribution. The type I receptor complex is expressed in many nonhepatic cell types (Doyle and others 2006), accounting for much of the hematologic and systemic toxicity observed with alfa interferons (Doyle and others 2006; Zhou and others 2007). Conversely, the type III receptor complex is expressed predominantly by hepatocytes and other epithelial or epithelial-like cells (Doyle and others 2006; Zhou and others 2007). These observations suggest that the 2 classes of interferons may differ with respect to safety and tolerability. Differences were confirmed in the EMERGE clinical trial, which demonstrated improved safety and tolerability of peginterferon lambda-1a (Lambda) versus peginterferon alfa-2a (Alfa) (Muir and others 2014b).
In addition to TVR and boceprevir, a number of direct-acting antiviral (DAA) treatments for chronic HCV have also been identified. One such DAA is daclatasvir (DCV), a potent, orally administered NS5A replication complex inhibitor (Herbst and Reddy 2013). Clinical trials have demonstrated that this agent is well tolerated, convenient, and associated with high sustained virologic response (SVR) rates when used with other agents in all oral regimens for the treatment of chronic HCV GT1b infection (Pol and others 2012; Everson and others 2014; Kumada and others 2014; Lok and others 2014; Manns and others 2014; Sulkowski and others 2014; Muir and others 2015; Poordad and others 2015).
Given the potential for improved efficacy and safety with Lambda and DCV compared with the former SOC, this clinical study (NCT01718158) was initiated to compare the efficacy and safety of lambda-1a combined with ribavirin/daclatasvir (Lambda/RBV/DCV) versus alfa-2a combined with ribavirin/telaprevir (Alfa/RBV/TVR) in patients with chronic HCV GT1b infection.
Materials and Methods
Trial design
This trial was a prospective, open-label, randomized phase 3 study conducted at 85 sites in 13 countries (Argentina, France, Germany, Israel, Italy, Japan, Korea, Poland, Russia, Spain, Taiwan, United Kingdom, and United States) between January 2013 and October 2014.
Patients were randomly assigned in a 2:1 ratio to receive Lambda/RBV/DCV or Alfa/RBV/TVR. Randomization was stratified according to interleukin-28B (IL28B) host GT (CC versus non-CC), treatment-naïve versus relapse status, and region (Japan versus rest of the world), and was performed by a sponsor-designated center via an Interactive Voice Response System using a block size of 6.
Patients in the Lambda/RBV/DCV arm initially received the combination of Lambda [180 μg subcutaneous (SC) injection once weekly], RBV (oral twice daily for a total daily dose of 1000–1200 mg), and DCV (60 mg tablet once daily) for 12 weeks, followed by an additional 12 weeks of Lambda/RBV, to complete a total of 24 weeks of therapy. Patients in the Alfa/RBV/TVR arm initially received the combination Alfa (180 μg SC injection once weekly), RBV, and TVR (two 375 mg tablets 3 times daily) for 12 weeks, followed by an additional 12 weeks of Alfa/RBV, after which patients received response-guided treatment based on extended rapid virologic response (eRVR) status as recommended by the Food and Drug Administration guidelines for TVR in treatment-naïve patients and prior relapsers (Liu and others 2013). Alfa/RBV was stopped in eRVR-positive patients; eRVR-negative patients received another 24 weeks of treatment. Patients in both arms were followed for 24 weeks after the end of treatment (EOT).
Treatment in the Lambda/RBV/DCV arm was discontinued at any time from week 4 if there was a virologic breakthrough [defined as a confirmed >1 log10 increase in HCV RNA over nadir or confirmed HCV RNA ≥ the lower limit of quantification (LLOQ) after previously having an HCV RNA < LLOQ while on treatment], or at week 12 if HCV RNA was >1000 IU/mL. As per the label, treatment in the Alfa/RBV/TVR arm was discontinued at week 4 or 12 if HCV RNA was >1000 IU/mL or at week 24 if HCV RNA was detectable.
Patients
The study included adults (aged ≥18 years) with chronic HCV GT1b (viral load ≥100,000 IU/mL), who were treatment naïve or prior relapsers to peginterferon Alfa/RBV therapy. Participants were required to have a positive anti-HCV antibody, HCV RNA, or a HCV GT test at least 6 months before screening, or a liver biopsy or FibroScan® (Echosens, Paris, France) consistent with chronic HCV infection (ie, evidence of fibrosis and/or inflammation). Patients with compensated cirrhosis without evidence of portal hypertension were also eligible for the study.
Patients infected with an HCV GT other than 1b, or who had hepatitis B surface antigen or a positive human immunodeficiency virus (HIV)-1/HIV-2 antibody test at screening, were excluded from the study. Other exclusion criteria were chronic liver disease unrelated to HCV, current evidence or known history of decompensated cirrhosis, and any condition that would contraindicate alfa interferon treatment. Patients with a hemoglobin concentration <12.0 g/dL (male) or <11.0 g/dL (female), a platelet count <90,000/mm3, or total serum bilirubin ≥2 mg/dL (unless due to Gilbert's syndrome) were also excluded.
Enrollment was capped at ≈10% for the number of patients with cirrhosis, ≈20% for the number of relapsers, and ≈20% for the number of Japanese patients.
This study was conducted in accordance with Good Clinical Practice defined by the International Conference on Harmonization and in accordance with the principles of the Declaration of Helsinki and underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). All patients provided written informed consent. The trial protocol, amendments, informed consent documents, and other materials provided to patients were approved by the appropriate independent ethics committee or institutional review boards for each participating site before study initiation.
Endpoints and assessments
The primary endpoint was the proportion of patients who achieved SVR at post-treatment follow-up week 12 (SVR12). The secondary efficacy endpoints were the proportion of treatment-naïve patients who achieved SVR12 and the proportion of patients who achieved SVR at post-treatment follow-up week 24 (SVR24).
Safety was assessed throughout the study based on AE and serious AE (SAE) reporting and clinical laboratory tests. Grading of AEs was based on the Division of AIDS table for Grading the Severity of Adult and Pediatric Adverse Events version 1.0 (2004; August 2009 clarification). Specific secondary endpoints relating to safety were the proportion of patients with rash-related dermatologic AEs during the first 12 weeks of treatment; treatment-emergent cytopenic abnormalities [anemia (hemoglobin <10 g/dL), and/or neutropenia (absolute neutrophil count <750/mm3), and/or thrombocytopenia (platelets <50,000/mm3)] through to EOT; flu-like symptoms (pyrexia, chills, or pain); or musculoskeletal symptoms (arthralgia, myalgia, or back pain) through EOT.
Efficacy and safety assessments were made at weeks 4 and 12 after treatment initiation. Follow-up assessments were performed at 4, 12, and 24 weeks post-treatment.
Statistical analysis
A 2-stage evaluation of SVR12 between Lambda/RBV/DCV and Alfa/RBV/TVR was planned. In the first stage, the noninferiority of Lambda/RBV/DCV to Alfa/RBV/TVR was tested. Provided noninferiority was established, a second stage test was conducted to demonstrate superiority.
With 300 patients treated with Lambda/RBV/DCV and 150 patients treated with Alfa/RBV/TVR, there was 95% power to demonstrate noninferiority of Lambda/RBV/DCV to Alfa/RBV/TVR for the proportion of patients with SVR12 at follow-up week 12. This was assuming an 85% response rate for both Lambda/RBV/DCV and Alfa/RBV/TVR and a 12% noninferiority margin for comparison with the lower limit of the 2-sided 95% confidence interval (CI) for the treatment difference (Lambda/RBV/DCV versus Alfa/RBV/TVR). This planned sample size would also provide 90% power for testing superiority of Lambda/RBV/DCV to Alfa/RBV/TVR assuming a 95% and 85% response rate for Lambda/RBV/DCV and Alfa/RBV/TVR, respectively, and a type I error of 0.05 (2-sided).
The modified intent-to-treat (mITT) approach was used for the analyses. Treatment difference in SVR12 rate and 95% CI were estimated using the stratum-adjusted Mantel-Haenszel approach. The treatment comparison for the secondary endpoints was performed only if the comparison of the primary endpoint (SVR12) demonstrated noninferiority. For treatment difference in SVR24 rate, observed values were used rather than mITT values. Given the evolving HCV treatment landscape and the approval of several DAAs with increased efficacy and tolerability profiles, the Lambda development program was terminated in the second half of 2014; therefore, follow-up after SVR12 was not completed in all subjects in this study. All testing was performed at a significance level of 0.05.
Results
Patient disposition and baseline characteristics
As shown in Fig. 1, a total of 444 patients were randomized and 440 patients initiated treatment, including 294 in the Lambda/RBV/DCV arm and 146 in the Alfa/RBV/TVR arm. The study was completed by 97.6% and 94.2% patients in the Lambda/RBV/DCV and Alfa/RBV/TVR arms, respectively. The proportion of patients completing treatment was 92.2% and 67.8% in the Lambda/RBV/DCV and Alfa/RBV/TVR arms, respectively. This difference between the 2 arms was mainly driven by a lower rate of discontinuations due to AEs in the Lambda/RBV/DCV arm (5.4%) than in the Alfa/RBV/TVR arm (19.2%).
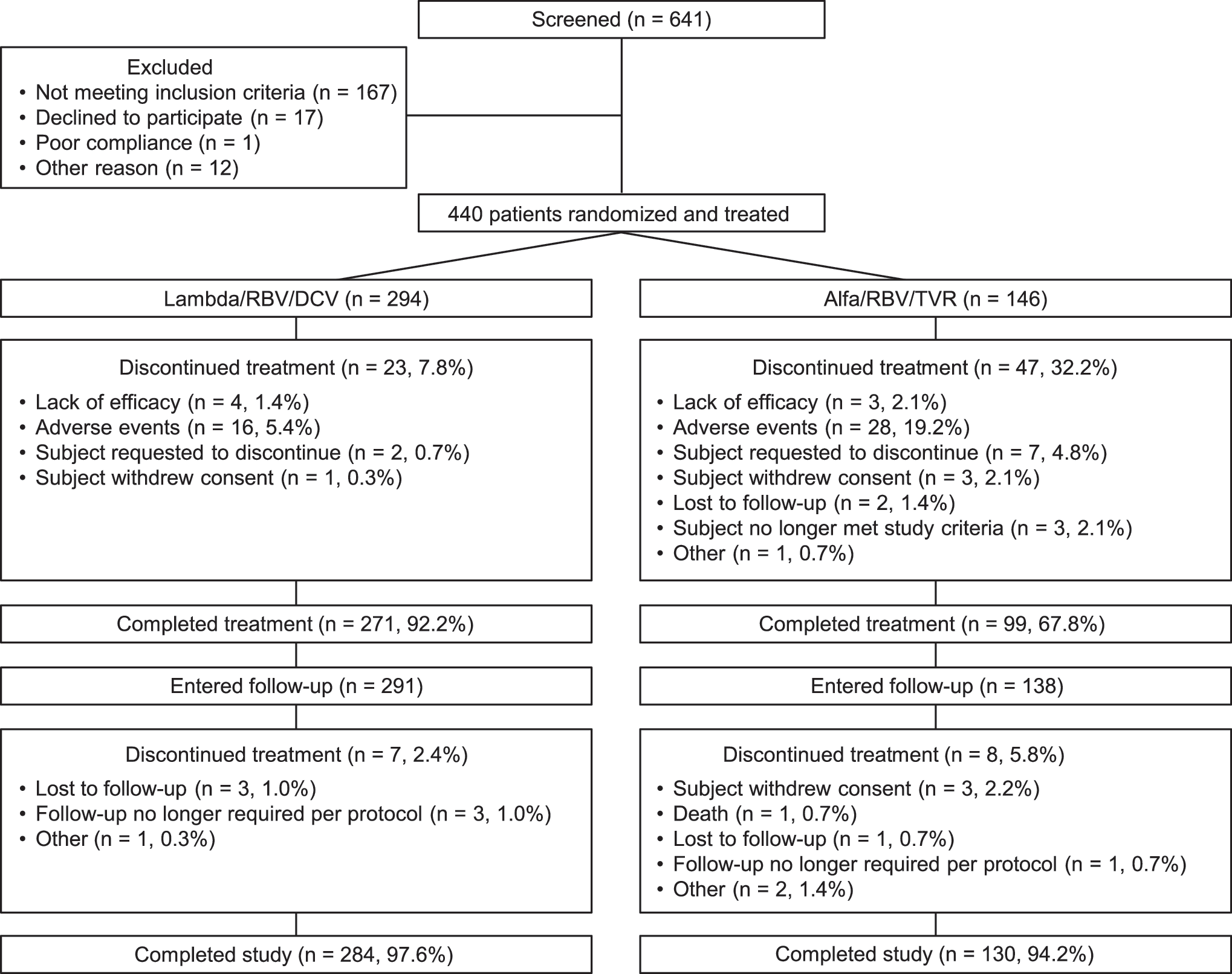
Patient disposition and reasons for discontinuation. Alfa, peginterferon alfa-2a; DCV, daclatasvir; Lambda, peginterferon lambda-1a; RBV, ribavirin; TVR, telaprevir.
Patient demographics and baseline disease characteristics were similar between the 2 study arms (Table 1). The majority of patients were Caucasian, treatment naïve, and ≈38% of patients had a CC IL28B GT.
Alfa, peginterferon alfa-2a; Alfa/RBV/TVR, alfa-2a combined with ribavirin/telaprevir; DCV, daclatasvir; GT, genotype; HCV, hepatitis C virus; IL28B, interleukin-28B; Lambda, peginterferon lambda-1a; RBV, ribavirin; TVR, telaprevir.
Efficacy
The proportion of patients achieving SVR12 (primary endpoint) was 88.8% (261/294; 95% CI: 85.2–92.4) in the Lambda/RBV/DCV and 70.5% (103/146; 95% CI: 63.2–77.9) in the Alfa/RBV/TVR arm. The difference between the Lambda/RBV/DCV arm and the Alfa/RBV/TVR arm was statistically significant (18.3%; 95% CI: 9.9–25.7; P < 0.0001) (Table 2 and Fig. 2A).
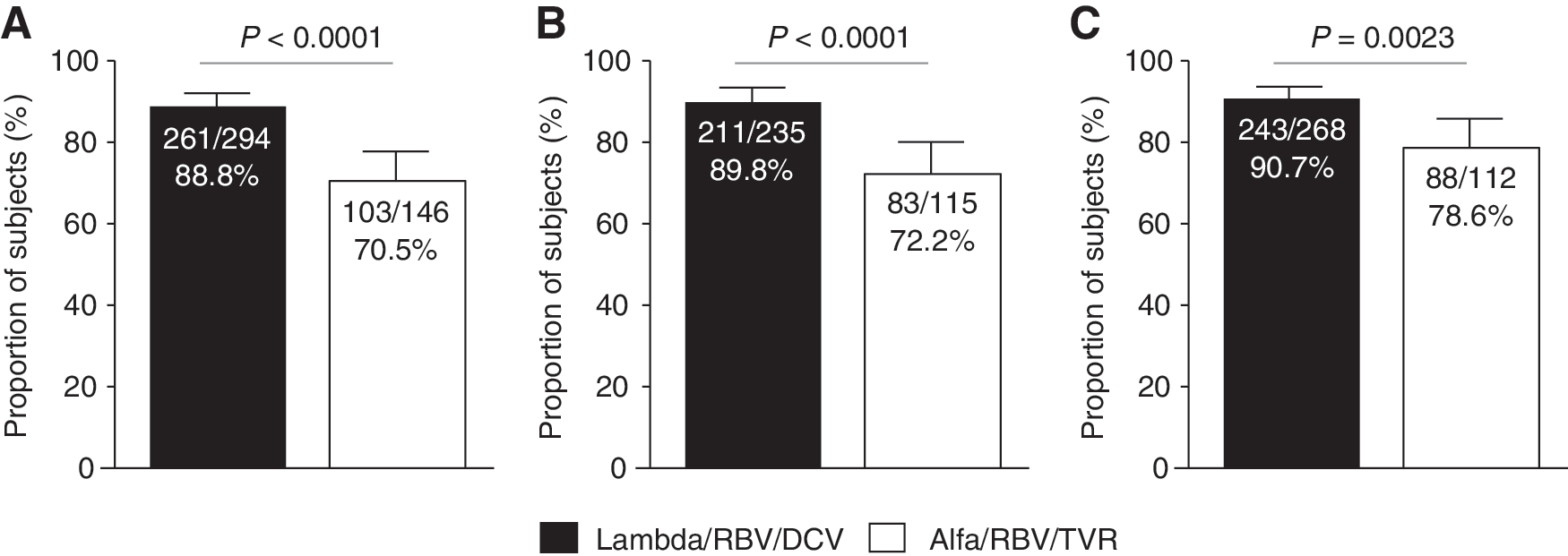
Proportion of patients with
Denominator is number of treated patients who had undetectable HCV RNA at EOT.
EOT, end of treatment; Lambda/RBV/DCV, lambda-1a combined with ribavirin/daclatasvir.
The proportion of treatment-naïve patients achieving SVR12 was 89.8% (211/235; 95% CI: 85.9–93.7) and 72.2% (83/115; 95% CI: 64.0–80.4) in the Lambda/RBV/DCV and Alfa/RBV/TVR arms, respectively. Again, the difference between the 2 arms was statistically significant (17.6%; 95% CI: 8.8–26.3; P < 0.0001) (Fig. 2B). The proportion of relapsers who achieved SVR12 was 83.9% and 66.7% in the Lambda/RBV/DCV and Alfa/RBV/TVR arms, respectively (not shown).
As mentioned previously, the study was prematurely terminated; therefore, SVR24 findings are expressed using observed values. The proportion of patients achieving SVR24 was 90.7% (243/268; 95% CI: 87.2–94.2) for the Lambda/RBV/DCV group and 78.6% (88/112; 95% CI: 71.0–86.2) for the Alfa/RBV/TVR group, with a significant difference between arms (12.1%; 95% CI: 4.3–20.0; P = 0.0023) (Fig. 2C).
The proportion of patients experiencing SVR12 virologic failure for any reason was 11.2% in the Lambda/RBV/DCV group and 29.5% in the Alfa/RBV/TVR group (Table 2). Overall, in the Lambda/RBV/DCV group, 5.8% of patients relapsed after achieving undetectable HCV RNA at EOT, 2.4% had virologic breakthrough, 1.4% had other on-treatment failures, and 1.4% did not have a HCV RNA value at follow-up week 12. In the Alfa/RBV/TVR group, 13.7% of patients relapsed after achieving undetectable HCV RNA at EOT, 6.2% had other on-treatment failures, 4.8% did not have a HCV RNA value at follow-up week 12, and 2.7% had virologic breakthrough (Table 2). Virologic resistance testing was not performed due to early termination of the study.
Safety
Fewer patients in the Lambda/RBV/DCV group than in the Alfa/RBV/TVR group reported rash-related dermatologic events (26.5% versus 37.0%; P = 0.0177), cytopenic abnormalities (10.2% versus 56.2%; P < 0.0001), and flu-like symptoms (9.9% versus 27.4%; P < 0.0001). The proportion of patients reporting musculoskeletal symptoms in the Lambda/RBV/DCV arm was not significantly different from that in the Alfa/RBV/TVR arm (17.7% versus 19.9%; P = 0.5840) (Table 3).
P = 0.0177; b P = 0.5840; c P < 0.0001; d P < 0.0001 for the difference between the 2 groups; ehemoglobin <100 g/L; fabsolute neutrophil count <0.75 × 109 c/L; gplatelets <50 × 109 c/L; hnumber of patients who had treatment grade 1/2 or grade 3/4 total bilirubin and had nonmissing on-treatment direct bilirubin results is used as the denominator for the percentage.
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; SAE, serious AE; TBILI, total bilirubin; ULN, upper limit of normal.
With respect to grade 3/4 AEs (Table 3), increased aspartate aminotransferase (AST), blood bilirubin, alanine aminotransferase (ALT) and gamma-glutamyltransferase were reported for 14 (4.8%), 7 (2.4%), 5 (1.7%), and 4 (1.4%) patients, respectively, in the Lambda/RBV/DCV group. These events had a lower incidence in the Alfa/RBV/TVR arm. In the Lambda/RBV/DCV group, the incidences of neutropenia, anemia, lymphopenia, thrombocytopenia, and leukopenia grade 3/4 AEs were nil or lower compared to the corresponding incidences for the Alfa/RBV/TVR group, which were reported in 21 (14.4%), 12 (8.2%), 5 (3.4%), 4 (2.7%), and 2 (1.4%) patients, respectively.
Overall, SAEs (Table 3) were reported in 11 (3.7%) patients in the Lambda/RBV/DCV group and 16 (11.0%) patients in the Alfa/RBV/TVR group. Jaundice SAEs affected 3 patients (1%) in the Lambda/RBV/DCV group and no patients in the Alfa/RBV/TVR arm. Rash, malaise, and acute renal failure affected 5 (3.4%), 2 (1.4%), and 2 (1.4%) patients in the Alfa/RBV/TVR group, respectively, and no patients in the Lambda/RBV/DCV arm.
A summary of changes in laboratory evaluations is provided in Table 3. The majority of laboratory abnormalities on-treatment were mild to moderate (grade 1/2). Hematological laboratory test abnormalities were less frequent in the Lambda/RBV/DCV group than in the Alfa/RBV/TVR group. These events were primarily responsible for an increased rate of RBV dose reductions in the Alfa/RBV/TVR group. Overall, 13.6% and 53.4% of patients in the Lambda/RBV/DCV group and the Alfa/RBV/TVR group, respectively, had the RBV dose reduced.
Liver laboratory test abnormalities were more frequent in the Lambda/RBV/DCV group than in the Alfa/RBV/TVR group (grade 3/4 AEs; see Table 3 for further details). However, no patient in the Lambda/RBV/DCV group met the laboratory criteria for potential drug-induced liver injury. In contrast, 1 patient in the Alfa/RBV/TVR group met these criteria. Elevated liver function tests led to Lambda dose reductions in 19 patients (6.5%) and to Alfa dose reductions in 1 patient (0.7%). Hepatic AEs accounted for AE-related discontinuations in 14 of 16 (88%) patients in the Lambda/RBV/DCV group and 2 of 28 (7%) patients in the Alfa/RBV/TVR group.
One patient in the Alfa/RBV/TVR group died of myocardial infarction during follow-up week 12, which was deemed by the investigator as unlikely to be drug-related.
Discussion
In this phase 3 study, the 24-week Lambda/RBV/DCV regimen was more effective than the Alfa/RBV/TVR combination in patients with HCV GT1b infection, irrespective of whether patients were treatment naïve or relapsers. A statistically significant difference was found between the Lambda/RBV/DCV and Alfa/RBV/TVR treatment groups for SVR12.
The EMERGE study previously demonstrated that Lambda/RBV and Alfa/RBV are similarly effective in patients with GT1 infection, with SVR12 rates of ≈40% (Muir and others 2014b). Therefore, the superior effectiveness of Lambda/RBV/DCV compared with Alfa/RBV/TVR in this study was most likely driven by the potency of DCV and the better tolerability of Lambda/RBV/DCV versus Alfa/RBV/TVR. The high SVR rate with the Lambda/RBV/DCV regimen in this study is consistent with those reported by other studies which combined DCV in an interferon-based regimen in similar patient populations (Lok and others 2012, 2014; Hezode and others 2015).
Lambda/RBV/DCV compared favorably with Alfa/RBV/TVR in terms of flu-like symptoms, hematologic AEs, and dermatologic AEs. In line with these observations, SAEs and discontinuations due to AEs were lower in the Lambda/RBV/DCV arm. Lower rates of flu-like symptoms and cytopenic events are consistent with the restricted distribution of Lambda receptors compared with the wider extrahepatic distribution of Alfa receptors (Lauer and Walker 2001; Doyle and others 2006; Zhou and others 2007). Lower rates of flu-like symptoms and cytopenic events observed in the Lambda/RBV/DCV arm corroborate the findings observed in previous clinical studies comparing these 2 agents (Muir and others 2013, 2014b).
Additionally, the lower rates of cytopenic and dermatologic events in the Lambda/RBV/DCV arm also reflect the improved safety profile of DCV versus TVR. Prior studies have shown that the addition of TVR to peginterferon alfa/RBV increases the rates of anemia and rash (Jacobson and others 2011; Zeuzem and others 2011), leading to a relatively high discontinuation rate (Colombo and others 2014). In contrast, rates of SAEs and treatment discontinuations previously have been shown to be similar between DCV and placebo when added to Alfa/RBV (Hezode and others 2015).
The Lambda/RBV/DCV regimen was associated with an increased rate of liver-related lab abnormalities (including increased bilirubin, ALT, and AST) compared with Alfa/RBV/TVR. This is in line with previous observations comparing Lambda versus peginterferon alfa (Muir and others 2013, 2014b). While the underlying mechanism responsible for the increased ALT and AST associated with interferons is largely unknown, similar but lower rates of increased hepatic transaminases have been observed with Alfa compared with Lambda (Muir and others 2014b).
All patients in the study were infected with HCV GT1b, which was previously shown to be one of the GTs most sensitive to DCV when combined with an interferon-based (SVR24: 76%–77%) or all-oral (SVR12: 82%–90%) regimen (Manns and others 2014; Hezode and others 2015). A higher genetic barrier to DCV resistance in GT1b patients accounts for the increased response rates observed in GT1b versus GT1a patients (Bunchorntavakul and Reddy 2015). Previous studies have shown that the rate of virologic failure due to emergence of DCV-associated resistant variants was higher in GT1a patients (Aghemo and De Francesco 2015; Hezode and others 2015).
Since this trial was initiated, all-oral regimens have been shown to be highly effective in patients with chronic HCV infection, including DCV-based regimens (Everson and others 2014; Kumada and others 2014; Lok and others 2014; Manns and others 2014; Sulkowski and others 2014; Muir and others 2015; Poordad and others 2015) (see Herbst and Reddy 2013, for review). Due to reduced treatment duration and all-oral administration, these regimens have improved tolerability and convenience, in addition to higher efficacy, compared with traditional interferon-based regimens. As a result, they have become the new SOC for most patients with HCV (Bunchorntavakul and Reddy 2015). DCV is now approved in the United States, Europe, Japan, and multiple countries across the Americas, Middle East, and Asia Pacific region (Bunchorntavakul and Reddy 2015). This study further establishes the utility of DCV in treating HCV GT1b-infected patients.
Conclusions
Lambda/RBV/DCV had superior efficacy, was better tolerated, and led to fewer discontinuations due to AEs, compared with Alfa/RBV/TVR for treatment of HCV GT1b-infected, treatment-naïve, and previously relapsed patients. The clinical development of Lambda has been discontinued in light of favorable data with all-oral combinations compared with interferon-based regimens, including improved efficacy, safety, and convenience. Oral DCV is now approved in many countries worldwide, and the current data further confirm the potency, tolerability, and safety of this agent in patients with HCV GT1b infection.
Footnotes
Acknowledgments
We would like to acknowledge Eric Vandeloise for his significant contributions to the execution of the study. We would also like to thank the many investigators who contributed time and effort to this study, as well as Ryan Woodrow and Dena McWain of Infusion Communications for medical writing support and editorial assistance. This analysis and writing and editorial support was fully funded by Bristol-Myers Squibb.
Author Disclosure Statement
R.F.: advisory board member: AbbVie, Bristol-Myers Squibb, Gilead, Janssen, Merck, Novartis, Roche; speaker: AbbVie, Bristol-Myers Squibb, Gilead, Janssen, Merck, Roche. O.Z.: advisory board member: Merck Sharp and Dohme; speaker: AbbVie, Biocad, Bristol-Myers Squibb, Janssen, Merck Sharp and Dohme, Novartis, R-Pharm. A.G.: advisory board member: Bristol-Myers Squibb; speaker: Bristol-Myers Squibb. J-H.K: consultant: AbbVie, Bristol-Myers Squibb, Gilead Sciences, Roche; speaker: Bristol-Myers Squibb, Gilead Sciences, Merck Sharp and Dohme, Roche. R.Z.: employee: Bristol-Myers Squibb. S.P.: employee: Bristol-Myers Squibb. Y.D.: employee: Bristol-Myers Squibb. D.X.: employee: Bristol-Myers Squibb. S.S.: employee: Bristol-Myers Squibb. All other authors have no competing financial interests.