
Abstract
Perturbations in myelopoiesis are a common feature in solid tumor biology, reflecting the central premise that cancer is not only a localized affliction but also a systemic disease. Because the myeloid compartment is essential for the induction of adaptive immunity, these alterations in myeloid development contribute to the failure of the host to effectively manage tumor progression. These “dysfunctional” myeloid cells have been coined myeloid-derived suppressor cells (MDSCs). Interestingly, such cells not only arise in neoplasia but also are associated with many other inflammatory or pathologic conditions. MDSCs affect disease outcome through multiple mechanisms, including their ability to mediate generalized or antigen-specific immune suppression. Consequently, MDSCs pose a significant barrier to effective immunotherapy in multiple disease settings. Although much interest has been devoted to unraveling mechanisms by which MDSCs mediate immune suppression, a large gap has remained in our understanding of the mechanisms that drive their development in the first place. Investigations into this question have identified an unrecognized role of interferon regulatory factor-8 (IRF-8), a member of the IRF family of transcription factors, in tumor-induced myeloid dysfunction. Ordinarily, IRF-8 is involved in diverse stages of myelopoiesis, namely differentiation and lineage commitment toward monocytes, dendritic cells, and granulocytes. Several recent studies now support the hypothesis that IRF-8 functions as a “master” negative regulator of MDSC formation in vivo. This review focuses on IRF-8 as a potential target suppressed by tumors to cripple normal myelopoiesis, redirecting myeloid differentiation toward the emergence of MDSCs. Understanding the bases by which neoplasia drives MDSC accumulation has the potential to improve the efficacy of therapies that require a competent myeloid compartment.
Introduction
A
Our research has led us to focus on the regulation of the transcription factor, interferon regulatory factor-8 (IRF-8) as a critical target in this process. IRF-8 has been known for many years as a crucial regulator for normal myelopoiesis (Tamura and others 2015). It is required as a positive regulator for lineage commitment of myeloid progenitors (i.e., granulocyte–monocyte progenitors; GMPs) to monocytes/macrophages and dendritic cells (DCs) (Tsujimura and others 2002; Tamura and others 2008; Becker and others 2012). These cells all serve as antigen-presenting cells (APCs) to activate T-cell immunity. Accordingly, loss of IRF-8 expression or function may inversely drive the emergence of dysfunctional myeloid cells that lack full APC capacity. Such defective APCs, in turn, would facilitate rather than inhibit tumor progression through inadequate support of adaptive immunity. Evidence supporting this hypothesis, that regulation of IRF-8 is central to MDSC accumulation in cancer, will be presented in 3 sections: (1) our current understanding of MDSC biology; (2) known roles for IRF-8 in the immune system; and (3) tumor-induced alteration of IRF-8 expression underlying MDSC accumulation. Altogether, our understanding of transcriptional regulation of myeloid differentiation under normal and pathologic conditions is likely to bear important basic and translational implications for unraveling fundamental challenges confronting cancer immunotherapy.
MDSCs play important roles in tumor progression
The identification of MDSCs in basic research has relied primarily on phenotypic surface markers analyzed by methods such as flow cytometry or immunohistochemistry, followed by morphologic assessment and assays to demonstrate suppressive function. In mouse models, the most simplified characterization of MDSCs is coexpression of the canonical CD11b and Gr-1 cell surface markers. Further refinement in this field has subdivided MDSCs into 2 major subsets, monocytic or granulocytic, reflecting differential expression of 2 distinct isoforms of Gr-1: Ly6C and Ly6G (Movahedi and others 2008; Youn and others 2008). Therefore, the monocytic subset (M-MDSC) is further defined as CD11b+Ly6ChiLy6G−, while the granulocytic subset (G-MDSC) is defined as CD11b+Ly6CloLy6G+. As suggested by their name, these subsets are thought to reflect 2 branches in MDSC development, and their relative proportion in various pathologic states appears to be dependent upon the dominant cytokines produced in that disease process. It is noteworthy that in a vast majority of mouse tumor models tested, the G-MDSC subset tends to dominate over the M-MDSC subset (Gabrilovich and others 2012; Talmadge and Gabrilovich 2013).
In contrast to murine MDSC definitions, the phenotype of human MDSCs is much more complex. The most comprehensive analysis in cancer patients demonstrates at least 3 major myeloid subsets, including those with monocytic or granulocytic features as well as those lacking any mature myeloid monocytic/granulocytic characteristics (Diaz-Montero and others 2009; Mandruzzato and others 2009; Rodriguez and others 2009; Gabitass and others 2011; Raychaudhuri and others 2011; Talmadge and Gabrilovich 2013; Yu and others 2013; Damuzzo and others 2015; Marvel and Gabrilovich 2015; Messmer and others 2015). Each subset shares a “core” phenotype, CD11b+CD33+, the latter of which is a pan myeloid marker, but differs in the expression of CD14 (a marker of monocytes), CD15 (a marker of granulocytes), and/or HLA-DR (a marker of APC competence). Thus, the monocytic subset is defined as CD14+CD15−HLA-DRlo; the granulocytic subset, CD14−CD15+HLA-DR−; and the most immature myeloid subset is negative for all 3 markers (i.e., CD14−CD15−HLA-DR−). Despite the complex phenotypic patterns observed among the various human myeloid subsets, they do share a prominent feature of murine MDSCs, which is their ability to suppress adaptive T-cell responses.
Although “MDSC-like” cells are typically found at low frequencies in normal settings (<4% in peripheral tissues of mice and even less in humans) (Youn and others 2008; Messmer and others 2015), they have been shown to greatly expand in pathologic settings. Nearly universally, patients with various malignancies display perturbations in normal myelopoiesis (Messmer and others 2015), characterized by increased MDSCs in the bone marrow, peripheral blood, and lymphoid tissues, as well as the tumor microenvironment, where they can constitute > 5% of the total cells within the tissue (Youn and others 2008). Accumulation of such MDSCs generally correlates with worse overall survival, which is largely attributed to their ability to suppress immune responses. Intriguingly, MDSCs accumulate not only in cancer but also in a range of inflammatory disorders or immunopathologies, including the following: graft-versus-host disease (Highfill and others 2010), psychological stress (Jin and others 2013), infection (e.g., viral, bacterial and parasitic) (Goh and others 2013; Ray and others 2013) and autoimmunity (Crook and Liu 2014).
Several excellent reviews describing the mechanisms by which MDSCs suppress T-cell responses as well as directly promote tumor progression through proangiogenic activities have been published (Ostrand-Rosenberg and Sinha 2009; Ostrand-Rosenberg 2010; Condamine and Gabrilovich 2011; Gabrilovich and others 2012; Ostrand-Rosenberg and others 2012; Marvel and Gabrilovich 2015; Ugel and others 2015). Because of these protumorigenic abilities, extensive research in both mouse and human systems has been devoted to developing ways to deplete or alter the function of MDSCs, particularly in combination with other therapies (Draghiciu and others 2015). Specific targeting of MDSCs for therapeutic purposes is a field still considered to be in its infancy; however, this particular aspect of MDSC biology is beyond the scope of this review. Instead, the focus of this review is to highlight how MDSCs might arise at a transcriptional level, which is poorly understood.
As mentioned earlier, the exact cellular composition of the resulting myeloid population depends upon the disease setting, with different subsets emerging (i.e., monocytic vs. granulocytic). The phenotypic and morphologic heterogeneity of the MDSC response appears to be largely influenced by the type and quantities of the tumor-derived factors (TDFs) the malignancy secretes. A range of TDFs, such as GM-CSF, G-CSF, IL-10, CSF-1, VEGF, IL-1, IL-6, and PGE2 have been implicated as regulators of MDSC development, mobilization, expansion, or function (Kusmartsev and Gabrilovich 2006; Waight and others 2011). Signaling pathways activated by these TDFs and their potential convergence on the transcription factor IRF-8 will be a major focus later in this review. Accordingly, the next section will review fundamental knowledge of IRF-8 to provide a framework as to why IRF-8 may be an unrecognized player in tumor-induced MDSC formation.
The multifunctional talents of IRF-8: from immunity to oncogenesis
IRF-8, formerly known as interferon consensus sequence binding protein (ICSBP), is a member of the IRF family of transcription factors (Driggers and others 1990). In contrast to most IRF family members expressed in various cell types, IRF-8 is largely restricted to cells of myeloid and lymphoid lineages, predominantly monocytes/macrophages, several DC subsets, and B cells (Taniguchi and others 2001; Tamura and others 2008; Wang and others 2014). IRF-8 can be active under basal conditions, but is also strongly induced by the inflammatory cytokine IFN-γ.
IFN-γ is a critical cytokine for the induction of adaptive immunity, including antitumor responses (Zaidi and Merlino 2011). Schreiber and colleagues revealed an indispensable role for the IFN-γ system in tumor immunology (Farrar and Schreiber 1993; Mumberg and others 1999; Shankaran and others 2001; Ikeda and others 2002; Dunn and others 2004a, 2005). Disruption of IFN-γ signaling pathways significantly increases susceptibility to chemically induced and spontaneous arising tumors. IFN-γ binding to its cognate receptor results in the activation of Janus Kinase (JAK)-1 and JAK2. JAK1/2, in turn, phosphorylates the IFN-γ receptor, creating a docking site for STAT1 and its consequent phosphorylation. Activated STAT1 then forms STAT1:STAT1 homodimers, enabling the complex to translocate to the nucleus to bind specific consensus motifs known as GAS (IFN-γ activation sequence) sites within the promoter regions of responsive target genes, including IRF-8.
The transcription factor IRF-8 is a 48-kDa, 424 amino acid protein that consists of 2 major domains: a well-conserved N-terminal DNA binding domain (DBD) and a more variable C-terminal IRF association domain (IAD), which allows for interaction with other transcription factors, such as IRF-1, IRF-2, IRF-4, or PU.1 (Tamura and others 2008, 2015). The formation of these heterocomplexes is necessary for driving many of the known functions of IRF-8, as evidenced by the genetically impaired BXH2 mice (Turcotte and others 2004). These mice possess a cytosine-to-thymidine point mutation within the IRF-8 IAD, which results in a loss of heterocomplex formation. Importantly, BXH2 mice display a myeloproliferative disorder that phenocopies that observed in IRF-8 global knockout mice (Turcotte and others 2004, 2005), which is described later in this review.
IRF-8 activity is also modulated by the action of various kinases (Sharf and others 1997). Phosphorylation of IRF-8 in its DBD blocks activity, while phosphorylation in its IAD promotes heterocomplex formation with other transcription factors. Once in the nucleus, IRF-8 heterocomplexes can either activate or repress the transcription of a variety of downstream target genes by engaging specific DNA elements or “consensus motifs” within promoter regions (Levi and others 2002). Several major IRF-8 responsive consensus motifs have been identified, including the following: ISRE (IFN-stimulated response element), GAS, EICE (Ets-IRF composite element), and IECS (IRF-Ets composite sequence) (Kanno and others 1993; Kuwata and others 2002; Tamura and others 2005b, 2008; Smith and others 2011). Generally, IRF-8 binding to ISRE sites leads to repression, while IRF-8 binding to GAS, EICE, or IECS sites leads to activation (Nelson and others 1993; Tamura and others 2008, 2015). The ability of IRF-8 to selectively engage these DNA-binding regions is thus heavily influenced by the sequence of the cis element and its interacting partners.
Collectively, IRF-8 influences diverse activities within the immune system. More precisely, as illustrated in Fig. 1, the biologic roles of IRF-8 encompass 3 major levels: (1) developmental, (2) functional, and (3) oncogenic. Much of what is known about these diverse IRF-8-mediated bioactivities is derived from observations of the global IRF-8 knockout (IRF-8−/−) mouse (Holtschke and others 1996). Novel tools such as cell-specific loss of IRF-8, as well as IRF-8 reporter strains are beginning to strengthen these early findings.
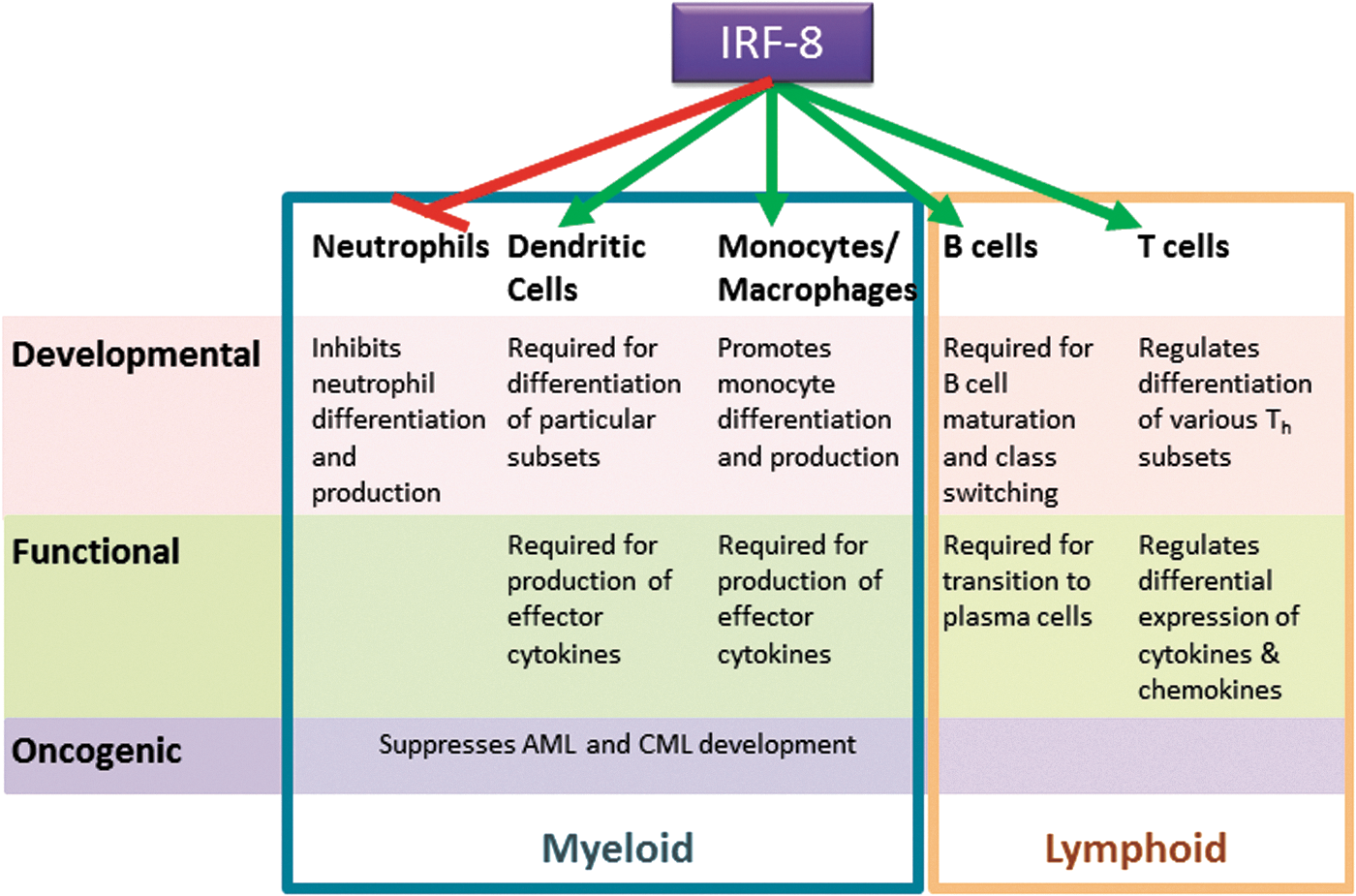
Multiple functions of IRF-8 in the immune system. Roles for IRF-8 in both the myeloid (blue box) and lymphoid (orange box) arms of the immune system can be stratified into 3 basic tiers reflecting 3 fundamental cellular processes: development (pink), function (green), and oncogenic potential (purple). IRF-8 either suppresses (red line) or supports (green lines) the various cell types and their activities, as indicated.
First, with regard to development, within the myeloid lineage steady-state or constitutive IRF-8 expression is well-characterized as a positive regulator monocytes and DC (particularly CD8α, plasmacytoid and CD103+ subsets); whereas, it simultaneously acts as a negative regulator of granulocytic differentiation, namely neutrophils (Tamura and others 2000, 2008; Tamura and Ozato 2002; Auffray and others 2009). The interplay between these 2 branches in myeloid development is the major focus of the latter half of this review. However, IRF-8 is also important in lymphoid development. IRF-8 regulates several genes important for B-cell development, including activation-induced cytidine deaminase (AICDA) (Lee and others 2006), a gene required for immunoglobulin class switching. Plasma cell maturation is also dependent on IRF-8, with IRF-8 binding to IRF-4 during the phase of pre-B-cell to B-cell transition (Lu and others 2003). IRF-8 also appears to be a negative regulator of Th17 differentiation (Ouyang and others 2011).
Second, with respect to function, IFN-γ-inducible IRF-8 expression is critical for adaptive immunity as it regulates the expression of a number of key cytokine genes in monocytes/macrophages/DCs, such as IL-12p40, IL-12p35, IL-18, and RANTES (Giese and others 1997; Kim and others 1999; Wang and others 2000; Masumi and others 2002; Liu and others 2004; Liu and Ma 2006). These inflammatory cytokines drive or facilitate NK, Th1, and CD8+ T-cell responses. IRF-8 expression within T cells can also modulate the cytokines and chemokines these effector cells produce (Lee and others 2015; Paschall and others 2015). Interestingly, T cells deficient for IRF-8 were found to increase the production of GM-CSF (Paschall and others 2015), a cytokine contributing to MDSC accumulation, which will be discussed later in this review, revealing a myeloid cell-extrinsic mechanism by which IRF-8 could regulate the MDSC response.
Third, with respect to oncogenesis, the complete absence of IRF-8, as in global IRF-8−/− mice, leads to profound neutrophilia. Over time, this myeloproliferative phenotype culminates in myeloid malignancies akin to chronic myeloid leukemia (CML) or acute myeloid leukemia (AML). Indeed, the original characterization of the IRF-8−/− mouse model revealed striking similarities to such human myeloid malignancies (Holtschke and others 1996). Substantially reduced IRF-8 mRNA levels have been reported in CML and AML patients, while recovery of IRF-8 expression has been associated with IFN-α-induced clinical responses (Schmidt and others 1998, 2001). Thus, IRF-8 has been strongly implicated as a tumor suppressor in certain hematopoietic cancers, such as CML and AML. Interestingly, genes already linked to leukemogenesis, such as Wilms' tumor gene 1 (WT1) and BCR-ABL, may work at least, in part, through IRF-8 based on the finding that both have been shown to downregulate IRF-8 expression (Vidovic and others 2010; Watanabe and others 2013).
The precise mechanisms by which IRF-8 suppresses myeloproliferative diseases, however, remain incompletely understood. Several key observations in myeloid cell biology have revealed that this activity is likely related to the ability of IRF-8 to regulate apoptosis. IRF-8 directly represses transcription of multiple antiapoptotic genes, notably Bcl-2, Bcl-XL, or FAP-1 (Fas-associated phosphatase-1) (Burchert and others 2004; Huang and others 2008; Hu and others 2013). Overexpression of IRF-8 enhances caspase-3 expression (Gabriele and others 1999). Overall, IRF-8 expression renders cells more susceptible to apoptosis by chemotherapeutics, irradiation, and death receptor (TRAIL and Fas ligand)-mediated pathways (Gabriele and others 1999; Liu and Abrams 2003; Burchert and others 2004; Greeneltch and others 2007; Huang and others 2008; Yang and others 2009; Hu and others 2013). IRF-8 can also activate the expression of genes with other tumor suppressor activities (e.g., PML, NF1, p15ink4b) (Schmidt and others 2004; Dror and others 2007; Huang and others 2007).
Thus, we have summarized a variety of functions carried out by IRF-8 in the immune system and the range of pathologic consequences that may emerge in its absence, from fundamental immune deficiencies to hematopoietic malignancies. The remainder of this review will focus more specifically on the role IRF-8 plays in modulating myeloid cell development and potential mechanisms by which this process may be undermined in cancer.
IRF-8 expression is indispensable for normal myeloid cell differentiation
IRF-8, in partnership with other transcription factors such as PU.1 as discussed earlier, is essential for normal myelopoiesis, the branch of hematopoiesis leading to competent monocytes, several DC subsets, and granulocytes. Several earlier studies suggested that during hematopoiesis, IRF-8 expression increases steadily with increasing lineage commitment up to the GMP stage (Tsujimura and others 2002). This progenitor represents the most proximal, upstream bifurcation point between monocytes/DCs and granulocytes in myelopoiesis. This has recently been confirmed by Morse and colleagues (Wang and others 2014) using a newly developed transgenic mouse, wherein EGFP is functionally knocked into the endogenous IRF-8 locus, generating an IRF-8-EGFP fusion protein that retains the transcriptional activity. Thus, anywhere IRF-8 is expressed, so too is EGFP. This study demonstrated expression of IRF-8 in the GMP, beyond which IRF-8 is retained in monocytes, but significantly diminished or lost in granulocytes.
Recently, a newly refined phenotypic definition of the GMP population was reported by Goodridge and colleagues (Yanez and others 2015) that challenged the role of IRF-8 in this population. Conventional analysis of the GMP by flow cytometry has shown bimodal expression of IRF-8, hinting at potential heterogeneity within the GMP (Yanez and others 2015). Using differential expression of the surface markers Ly6C and CD115, Goodridge and colleagues identified 3 distinct subsets within this conventionally characterized GMP population. Confirmed by colony-forming assays, they defined the most “oligopotent” GMPs as Ly6C−. Ly6C+ cells were found to have differential expression of the monocytic marker, CD115. In colony-forming assays, Ly6C+CD115+ cells produced primarily monocytic cells, while Ly6C+CD115− cells produced primarily granulocytes. Thus, these populations were defined as more lineage-committed progenitors, monocytic progenitors (MPs), and granulocytic progenitors (GPs). Interestingly, while the conventional or “total” GMP population is heterogeneous for IRF-8 expression, IRF-8 levels were found to be relatively low in the oligopotent GMPs, but increased dramatically after lineage commitment in both GPs and MPs. How IRF-8 levels increase from IRF-8lo oligopotent GMPs to IRF-8hi GPs and IRF-8hi MPs remained undefined. Thus, this study suggests that IRF-8 functions downstream of oligopotent GMPs to control monocyte and neutrophil production from MPs and GPs. We will discuss this new model further in the last section of this review and how it pertains to the origin of the MDSC response in cancer.
The essential role of IRF-8 in regulating the bifurcation point in myeloid cell differentiation was unveiled in IRF-8−/− mice. IRF-8−/− mice have profound changes in myelopoiesis with marked increases in the frequency of granulocytes (mainly neutrophils) in the bone marrow and periphery at the expense of monocytes (mainly Ly6C+) and several DC subsets, namely plasmacytoid, CD8α+, and nonlymphoid CD103+ (Schiavoni and others 2004; Tamura and others 2005a). As discussed earlier, a significant portion of mice develop a CML-like syndrome that ultimately progresses to a fatal blast crisis. Lethally irradiated wild-type mice reconstituted with bone marrow from IRF-8−/− mice go on to develop leukemia, demonstrating that the IRF-8 defect principally affects and originates within the hematopoietic compartment (Scheller and others 1999). Moreover, humans that harbor point mutations (i.e., K108E or T80A) in the DBD of IRF-8 also lack several DC subsets, as well as circulating monocytes, and have a reciprocal profound neutrophilia (Hambleton and others 2011). Thus, defects in myelopoiesis in humans with IRF-8 mutations essentially mimic what was observed in IRF-8−/− mice.
Importantly, myeloid progenitor cells from IRF-8−/− mice develop into granulocytes regardless of the type of myelopoietic growth factor (i.e., G-CSF, GM-CSF, or M-CSF) the cells are exposed to (Scheller and others 1999). Thus, IRF-8 is required for transition into competent monocytes; alternatively, IRF-8 suppresses the granulocytic transcriptional program, which is critical to control the size of the resultant neutrophil pool. Signaling by these cytokines has been tied to the regulation of IRF-8 expression and its binding partners. Later in this review, we will discuss these signaling pathways and their ability to modulate the outcome of myelopoiesis in normal and pathologic settings. First, however, we will discuss more key evidence for the connection between IRF-8 downregulation and MDSC production in cancer.
IRF-8 expression is reduced in myeloid cells in cancer
We have noted several commonalities between myeloid populations observed in IRF-8−/− mice and those present in tumor-bearing mice studied in our laboratory (Waight and others 2013). IRF-8−/− mice display significant splenomegaly due, in large part, to the accumulation of CD11b+Gr-1+ myeloid cells, with an approximately 7-fold enrichment of the granulocytic phenotype. These cells display T-cell suppressive activity consistent with G-MDSCs derived from tumor-bearing mice, and differential gene expression analysis revealed that the CD11b+Gr-1+ cells from IRF-8−/− mice were significantly more related to CD11b+Gr-1+ cells from tumor-bearing mice than those of the nontumor-bearing control group (Waight and others 2013). Interestingly, when IRF-8 expression was enforced in myeloid cells under control of the CD11b promoter (using a transgenic mouse model), the frequencies of MDSCs measured in the spleen, bone marrow, and tumor microenvironment of mouse tumor models were significantly decreased, with the largest decline observed in the G-MDSC subset (Waight and others 2013).
In a subsequent study (Paschall and others 2015), it was demonstrated that MDSCs arise not only from an intrinsic mechanism, but also an extrinsic mechanism of IRF-8 regulation. Interestingly, IRF-8-deficiency in T cells was found to contribute to MDSC accumulation and did so indirectly through a GM-CSF-dependent mechanism. Proof-of-concept experiments revealed that treatment of mice with recombinant GM-CSF increased MDSC accumulation, and the adoptive transfer of IRF-8 deficient, but not GM-CSF-deficient T cells, increased MDSC accumulation in the recipient hosts. Thus, these additional data supported a second model whereby IRF-8 loss in T cells facilitated MDSC generation indirectly through increased GM-CSF production. Additional experiments (Waight and others 2013) showed that GM-CSF can downregulate IRF-8 expression in myeloid populations through a STAT5-dependent mechanism. Taken collectively, these studies support an integral role of IRF-8 in the mechanism of MDSC development/expansion, which may occur through either intrinsic or extrinsic mechanisms.
We extended these studies to patient samples to evaluate the importance of IRF-8 expression in human MDSC biology (Waight and others 2013), focusing on the most immature MDSC population defined as CD33+CD14−CD15−HLA-DR−. Our analysis of peripheral blood leukocytes from breast cancer patients with advanced invasive ductal carcinoma (stage III/IV) revealed a significant increase in the percentage of this MDSC subset compared to female age- and race-matched controls. Initially, no significant difference in IRF-8 expression was observed between healthy donors and patients. However, when IRF-8 expression was plotted in relation to the percentage of MDSCs within the patient peripheral blood, a significant negative correlation was observed. No inverse correlation was observed for the controls, suggesting that the inverse relationship between MDSC frequency and IRF-8 status was disease dependent. Thus, in breast cancer patients, decreasing IRF-8 expression is associated with increased MDSC frequency. Importantly, MDSC frequencies have clinical significance, such that stratification of patients based on MDSC burden showed significant increases in progression-free and overall survival when MDSC frequencies were low.
Transcriptional regulation of MDSC development: signaling pathways that perturb IRF-8 function
Based on the key findings that IRF-8 deficiency generates a population of cells bearing a striking resemblance to G-MDSCs, we hypothesized that tumor-induced downregulation of IRF-8 expression in myeloid progenitors explains the phenotypic properties and robust expansion of these cells in the cancer setting. This raised the fundamental question addressed in this section: What tumor- or stromal-derived factors regulate IRF-8 to initiate this G-MDSC production? Refer to Fig. 2 for an outline of the critical signaling pathways described below.
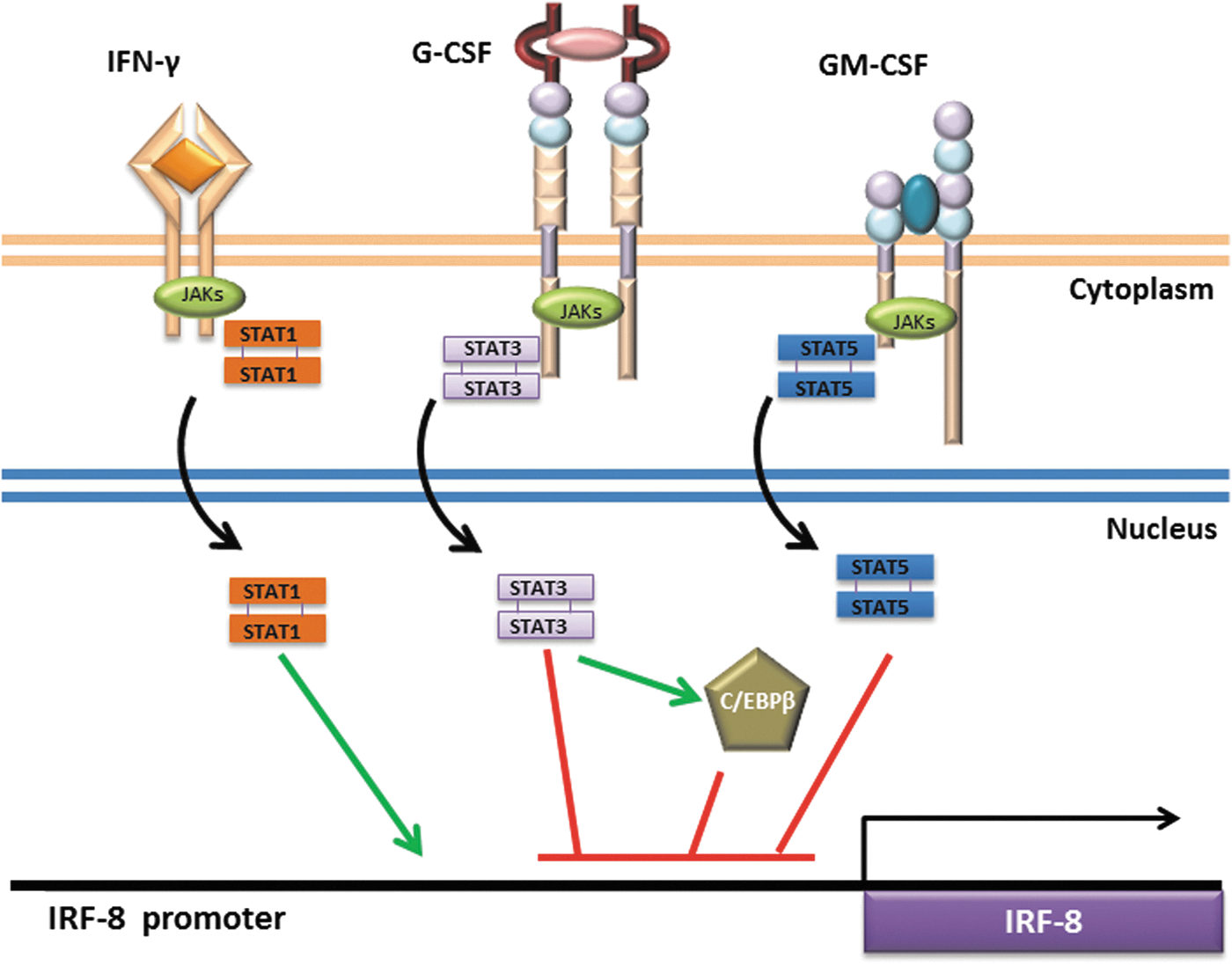
Signaling pathways regulating IRF-8 expression. Cytokines signal through engagement of specific cell surface receptors on myeloid populations. The major pathways discussed are highlighted, which illustrate the ability of IFN-γ to induce IRF-8 (green line), or G-CSF or GM-CSF to suppress IRF-8 (red lines) expression. The green or red lines shown in the nucleus are used to simply illustrate the concept that the different transcription factors inhibit IRF-8 transcription, not necessarily the precise location of their binding elements within the IRF-8 promoter. STATs are activated by phosphorylation by JAKs and subsequent translocation into the nucleus (black arrows). STAT3 induces (green arrows) the transcription factor C/EBPβ, which may also negatively regulate IRF-8 (at least in DC biology). In addition, cytokine signaling induces mechanisms of feedback inhibition, such as regulation of STAT1 by SOCS1, STAT3 by SOCS3, or STAT5 by SOCS2 (not shown).
The cytokine G-CSF is critical for both steady-state (homeostatic) granulopoiesis and a state of “emergency” granulopoiesis (Panopoulos and Watowich 2008; Manz and Boettcher 2014). Emergency granulopoiesis describes the massive production of granulocytes from the bone marrow in response to circulating G-CSF secreted by both hematopoietic and nonhematopoietic cell types during an acute systemic infection. However, given the rapid kinetics and robustness of this myeloid response, not all cells mature appropriately or at the same rate. Therefore, this response is characteristically heterogeneous, comprising neutrophils reflecting diverse (early and late) stages of differentiation (Manz and Boettcher 2014).
G-CSF is also produced by certain solid cancer types (Tsukuda and others 1993; Kyo and others 2000; Savarese and others 2001; Chakraborty and Guha 2007; Joshita and others 2009; Waight and others 2011; Aliper and others 2014) and as a single myelopoietic growth factor can proficiently drive systemic G-MDSC accumulation. Interestingly, the magnitude and characteristics of the resulting tumor-induced G-MDSC response is phenotypically, functionally, and molecularly similar to that induced by recombinant G-CSF administration (Waight and others 2011), another approach to initiate or mimic emergency granulopoiesis. The difference between G-CSF administration (or an acute inflammatory response) and cancer, however, is that the granulocytic outburst during the former process is transient and self-limiting, whereas the granulocytic outburst during the latter process is prolonged because of continued neoplastic exposure.
Following G-CSF engagement with its receptor, JAK1, JAK2, and TYK2 are recruited to the receptor, which in turn leads to the phosphorylation and activation of STAT3 (signal transducer and activator of transcription-3) (Tian and others 1994; Panopoulos and Watowich 2008). STAT3 is one of the most prevalent STATs associated with MDSC biology (Kujawski and others 2008; Poschke and others 2010; Rebe and others 2013; Condamine and others 2015). Tumor-infiltrating MDSCs display high levels of activated STAT3 (Nefedova and others 2005; Rebe and others 2013). Inhibition of STAT3 activity in vitro or in vivo results in reduced MDSC accumulation (Kortylewski and others 2005; Xin and others 2009; Tu and others 2012; Condamine and others 2015). For example, the ability to generate MDSCs in vitro from hematopoietic progenitor cells that express a dominant-negative retroviral STAT3 construct was strongly blocked (Nefedova and others 2004). Conversely, the overexpression of activated STAT3 in these myeloid progenitors promoted MDSC accumulation.
Many additional TDFs (including GM-CSF, VEGF, IL-6, and IL-10) also signal through STATs. Thus, even in cancer settings where G-CSF is not overexpressed, STAT family members can play a central role in the biology of MDSCs. As we will discuss, STAT5 activation downstream of GM-CSF is also implicated in MDSC development (Waight and others 2013). We and others have shown that STAT3 and STAT5 downstream of G-CSF and GM-CSF engagement act as negative regulators of IRF-8 transcription (Esashi and others 2008; Waight and others 2013). Exposure of bone marrow-derived myeloid cells to either cytokine significantly reduces IRF-8 expression within 2–3 h following treatment (Waight and others 2013). STAT3 and STAT5 inhibitors, FLLL32 (Bill and others 2012) and pimozide (Nelson and others 2011), respectively, were able to block this IRF-8 downregulation by G-CSF or GM-CSF. Chromatin immunoprecipitation assays (ChIP) provided direct evidence for binding of activated STAT3 or STAT5 to the IRF-8 promoter.
Moreover, we have identified an unrecognized role for STAT5 repression of IRF-8 in human and murine myeloid leukemias (Waight and others 2014). Activation of STAT5 downstream of the oncogenic BCR-ABL fusion protein directly represses IRF-8 transcription. Establishing a STAT5-IRF-8 axis in BCR-ABL-induced leukemias not only expands our understanding of the signaling pathways involved in the pathogenesis of such a myeloid disease but also helps to explain why IRF-8−/− mice develop a CML-like disease in the absence of BCR-ABL. Although the mechanisms are different, one BCR-ABL driven and the other a genetic deletion, the outcome is the same, which results in IRF-8 loss.
Cytokine-driven cellular activation is typically followed by feedback inhibition to ensure cellular homeostasis, particularly after the pathologic insult has been cleared or the inflammatory episode has been silenced. To that end, STAT activation in response to cytokine stimulation drives transcription of genes that act as negative regulators of signaling, known as suppressors of cytokine signaling (SOCS) (Kubo and others 2003). In the case of G-CSF signaling, the major negative regulator induced is SOCS3, which blocks G-CSF-induced signal transduction by binding to the G-CSF receptor (Hortner and others 2002). Yu and others reported that SOCS3-deficient bone marrow-derived myeloid cells are hyperresponsive to G-CSF, as measured by increased duration and intensity of G-CSF-induced STAT3 phosphorylation, which led to MDSC accumulation (Yu and others 2015). MDSCs from tumor-bearing SOCS3-deficient mice exhibited a greater ability to inhibit the T-cell activity. These findings highlight the critical role of G-CSF in MDSC biology and the importance of SOCS3 as a key negative regulator of this process. It is interesting to note that IRF-8−/− myeloid populations are hyperresponsive to cytokines such as G-CSF and GM-CSF, although the molecular basis for this hyperresponsiveness remains unknown, as well as whether there is cross-talk between IRF-8 and SOCS3.
STAT signaling also impacts the expression of potential binding partners of IRF-8. During emergency granulopoiesis, G-CSF-mediated STAT3 phosphorylation drives transcription of the CCAAT/enhancer-binding protein-beta (C/EBPβ) protein, a “master” regulator of emergency granulopoiesis (Hirai and others 2006). In contrast, C/EBPα expression is required for steady-state granulopoiesis (Zhang and others 1997; Radomska and others 1998). Transcription of C/EBPβ in cooperation with STAT3 drives MYC expression, which enables accelerated cell cycle progression and, consequently, heightened neutrophil output (Zhang and others 2010). Work by Bronte and colleagues defined a causal link between STAT3 and C/EBPβ expression in MDSC formation (Marigo and others 2010; Zhang and others 2010; Sonda and others 2011). A direct relationship between IRF-8 and C/EBPβ in G-MDSC formation, if any, remains to be investigated. However, a recent study demonstrated that IRF-8 and C/EBPβ antagonize each other and this relationship dictates the balance between 2 distinct DC subsets: plasmacytoid versus monocyte derived (Bornstein and others 2014). Thus, whether such an IRF-8-C/EBPβ axis may also exist in MDSC-tumor biology remains to be determined.
It is important to note that additional TDF-mediated signaling pathways may converge on and suppress expression of IRF-8 to generate MDSCs. Studies by Lyden and colleagues (Papaspyridonos and others 2015) found that IRF-8 was decreased following induction of the transcription factor Id1, downstream of TGF-β signaling in the setting of melanoma. Reconstitution of mice with bone marrow overexpressing Id1 generated a similar phenotype as IRF-8−/− mice, producing splenomegaly and functionally suppressive myeloid cells. However, whether Id1 directly binds the IRF-8 promoter to repress its expression or acts through an indirect mechanism has not yet been determined.
Expression of IRF-8 by myeloid progenitors is predicted to be a key molecular target underlying MDSC development
Based on the evidence discussed, we have developed a model for the generation of MDSCs in cancer illustrating where the suppression of IRF-8 expression/function may be contributing to their development (Fig. 3 for details). As described, circulating TDFs with hematopoietic growth factor-like characteristics are thought to initiate the sequence of events driving aberrant myelopoiesis. In malignancies where G-MDSCs dominate, we propose that aphysiologic levels of tumor-derived G-CSF constitute a relevant myelopoietic growth factor, which triggers STAT3 activation in myeloid progenitors. Activated STAT3 then translocates to the nucleus where it blocks IRF-8 transcription among other myelopoietic target genes, thereby skewing myeloid cell differentiation toward G-MDSC production. As noted, G-MDSCs dominate over M-MDSCs in many tumor models tested. However, numerous TDFs have been identified in a variety of tumor settings, which may differentially modulate MDSC biology. The contribution of IRF-8 suppression to the generation of M-MDSCs in such cancer models remains to be investigated.

Model for tumor-mediated suppression of IRF-8 during the production of G-MDSCs. Tumor-derived factors (TDFs) acting through STAT3, STAT5, or Id1 (for example) suppress IRF-8 transcription/expression during myeloid differentiation or at other cellular levels, which influence survival/persistence. This may occur in the bone marrow at the oligopotent granulocyte–monocyte progenitor (GMP) stage or the granulocytic progenitor (GP) stage, or in the periphery to drive expansion or enable persistence of circulating G-MDSCs (indicated by dashed red line). How potential loss of IRF-8 in monocytic progenitors (MPs) impacts monocyte production or M-MDSC accumulation in cancer requires further investigation.
As discussed earlier, the major function of IRF-8 in myeloid development or differentiation was originally thought to be at the GMP stage, a bifurcation point that oversees lineage commitment and the consequential production of monocytes versus granulocytes. However, the recent study by Goodridge and colleagues (Yanez and others 2015) offers new insights into the precise role played by IRF-8 to impact myeloid differentiation. This is due, in large part, to a refined definition of the GMP, which now divides the conventional or “total” GMP population into 3 separate progenitor populations, consisting of oligopotent GMPs and later stage GPs and MPs. While IRF-8 levels are low in oligopotent GMPs, IRF-8 levels are high in both GPs and MPs. Additional studies in IRF-8−/− mice revealed that the complete absence of IRF-8 did not impact GP or MP production, suggesting that their production is IRF-8 independent. Together with observations made in IRF-8−/− mice, it was then proposed that IRF-8 acts at the GP or MP stage and controls the fate of neutrophil or monocyte production at those stages. IRF-8 may do so by regulating lineage-specific myeloid programs, as well as signals associated with survival/apoptosis. Consequently, in order for monocytes to arise from MPs, IRF-8 levels remain high, whereas, for granulocytes to arise from GPs, IRF-8 levels decline.
Thus, in tumor-bearing hosts, we hypothesize that GPs would rise relative to MPs due to a corresponding loss of IRF-8 expression. The inverse relationship between GPs and their IRF-8 levels would provide a causal basis for the overabundance of G-MDSCs appearing in the periphery. Alternatively, the ability to upregulate IRF-8 expression during transition from oligopotent GMPs to GPs or MPs may be blocked. Interestingly, the levels of circulating GMPs, as currently defined in humans, have been shown to have a granulocytic bias and correlate with poorer prognostic features (Wu and others 2014).
While this review focused on the role and relevance of IRF-8 in MDSC biology, events downstream of IRF-8 still remain ill-defined. IRF-8 has been recently demonstrated to inhibit C/EBPα activity (Kurotaki and others 2014), a master regulator of steady-state granulopoiesis (Zhang and others 1997; Radomska and others 1998), thereby restricting neutrophil numbers. This antagonism may also be active in the production of G-MDSCs. On the other hand, in keeping with the idea that G-MDSC accumulation shares a similar signaling pathway with emergency granulopoiesis, it is also possible that an IRF-8-C/EBPβ axis may exist, which warrants further investigation. Our model also allows for the possibility that IRF-8 suppression in the periphery may be a contributing factor to G-MDSC accumulation. Treatment in vitro of bone marrow derived CD11b+ cells, a relatively well-differentiated myeloid population, with G-CSF rapidly reduced IRF-8 expression (Waight and others 2013). As IRF-8 is also required for the expression of genes associated with myeloid functionality, suppression of IRF-8 within intratumoral myeloid populations may contribute to functional MDSCs. Through the use of a genetic gain-of-function approach, we have also shown that enforced expression of IRF-8 under the CD11b promoter results in reduced MDSC numbers, namely G-MDSCs, under tumor-bearing conditions (Waight and others 2013). These data suggest that IRF-8 may also modulate G-MDSCs numbers in the periphery by influencing susceptibility to apoptosis, which requires further study. While it remains uncertain exactly where IRF-8 downregulation may occur, what is clear is that for G-MDSCs to arise, IRF-8 levels must be restrained and kept very low, a notion that is consistent with IRF-8 downregulation during normal neutrophil differentiation and production.
Conclusions
Among the prominent mechanisms known to impede antineoplastic responses is the accumulation of immune suppressive/protumorigenic myeloid populations, termed MDSCs. The identification of IRF-8 as an important regulator of fundamental facets of MDSC biology offers new insights into the long-standing conundrum of how cancers perturb myelopoiesis. Moreover, new knowledge of IRF-8 in myeloid-tumor cell biology may provide sound molecular rationale for the manipulation of these cells to improve cancer immunotherapy protocols. Defining the upstream regulators of IRF-8 expression such as JAK1/JAK2, STAT3, STAT5, and Id1 suggests that drugs which block these molecules could restore IRF-8 expression to restrain MDSC production. In addition, therapeutic targeting of key elements downstream of the IRF-8 signaling pathway could influence MDSC fate and disrupt MDSC-mediated mechanisms of tumor progression. These studies can be further extended to myeloproliferative pathologies, such as CML, where IRF-8 is also a relevant molecular player of disease outcome.
Footnotes
Acknowledgments
SIA was supported by NIH/NCI grants, R01CA140622 and R01CA172105; Department of Defense, W81XWH-11-1-0394; and CSN was supported by NIHT32CA085183.
Author Disclosure Statement
Author declares no competing financial interests.