
Abstract
The interferon regulatory factor (IRF) family consists of transcriptional regulators that exert multifaceted and versatile functions in multiple biological processes. Their crucial role as central mediators in the establishment and execution of host immunity in response to pathogen-derived signals downstream pattern recognition receptors (PRRs) makes IRFs a hallmark of the host antiviral response. They function as hub molecules at the crossroad of different signaling pathways for the induction of interferon (IFN) and inflammatory cytokines, as well as of antiviral and immunomodulatory genes even in an IFN-independent manner. By regulating the development and activity of immune cells, IRFs also function as a bridge between innate and adaptive responses. As such, IRFs represent attractive and compulsive targets in viral strategies to subvert antiviral signaling. In this study, we discuss current knowledge on the wide array of strategies put in place by pathogenic viruses to evade, subvert, and/or hijack these essential components of host antiviral immunity.
Introduction
T
These pathways are triggered when conserved microbial structures of pathogens, referred as pathogen-associated molecular patterns (PAMPs), are detected by one of a variety of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), which are found on the cell surface or on endocytic compartments, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RNA-binding RIGI-like receptors (RLRs), and a growing number of DNA sensors located in the cytoplasm (Sancho and Reis e Sousa 2012; Broz and Monack 2013; Mansur and others 2014; Wu and Chen 2014; Motta and others 2015; Pandey and others 2015; Sparrer and Gack 2015; Yoneyama and others 2015).
Upon PAMP ligation, receptors dimerize and move to signaling sites where they associate with adaptor proteins to recruit signaling adaptors, forming higher order multiprotein supramolecular complexes that transmit signals from activated receptor to cellular response (Wu 2013; Kagan and others 2014; Brubaker and others 2015).
A major component of this response is transcriptional and results in the production of interferons (IFNs) and proinflammatory cytokines. Then IFNs, in an autocrine and paracrine manner, stimulate hundreds of genes involved in antimicrobial defense within distinct functional categories, including cell migration, metabolic reprogramming, tissue repair, and regulation of adaptive immunity (Rusinova and others 2013; Schneider and others 2014). These genes are coordinately and temporally regulated through the differential expression and post-translational modification (PTM) of a relatively small number of transcription factors that act together with transcriptional coregulators and chromatin-modifying complexes within a stimulated cell (Tjian and Maniatis 1994; Agalioti and others 2000).
Among these transcription factors and in addition to the prototypical and most extensively studied nuclear factor-κB (NF-κB) (Dev and others 2011), interferon regulatory factors (IRFs) represent essential regulators of the innate and adaptive immunity associated with host resistance against pathogens, including viruses. IRFs function as hub molecules that act at the crossroad of different signaling pathways to stimulate the expression of IFNs and inflammatory cytokines, as well as of a subset of IFN-stimulated genes (ISGs), even in an IFN-independent manner (Honda and Taniguchi 2006; Tamura and others 2008; Battistini 2012; Ikushima and others 2013; Collins and Mossman 2014). Some IRFs are themselves ISGs that function to amplify and maintain the IFN response. By regulating the development and activation of immune cells, IRFs also instruct adaptive immunity (Ikushima and others 2013; Ysebrant de Lendonck and others 2014).
The IRF family presently comprises 9 mammalian members (IRF1–IRF9) coded by distinct, but related, genes. They were initially identified as transcriptional regulators of type I IFN and ISGs (Honda and others 2006), but it was suddenly recognized that through the activation or repression of a variety of target genes, they exert a broad range of activities in different biological processes not only limited to regulation of immunity and inflammation in response to PRR but also linked to oncogenesis and metabolism even in an IFN-independent manner (Fig. 1), as reviewed extensively elsewhere (Taniguchi and others 2001; Takaoka and others 2008; Tamura and others 2008; Battistini 2009, 2012; Savitsky and others 2010; Collins and Mossman 2014; Zhao and others 2015). Their distinct and not overlapping functions may depend on slightly different DNA binding specificities within the broad IRF consensus sequence, patterns of expression, and/or association with other regulators and coregulators in transcription. This association is mediated by the IRF-associated domains (IAD) 1 and 2 identified within the carboxy-terminal region of IRFs and may determine specificity of DNA binding and whether the protein complex functions as a transcriptional activator or repressor (Taniguchi and others 2001; Tamura and others 2008).

The IRF family members. The word cloud contains a list of the broad range of IRF activities that highlight the essential roles of these transcription factors in immunity, oncogenesis, and metabolism. IRF, interferon regulatory factor. Color images available online at
Each family member, with the exception, so far, of IRF2 and IRF6, has been involved in antiviral signaling downstream PRRs (Fig. 2), with their contribution being nonredundant, cell-type, and stimulus specific (Honda and Taniguchi 2006; Ikushima and others 2013).
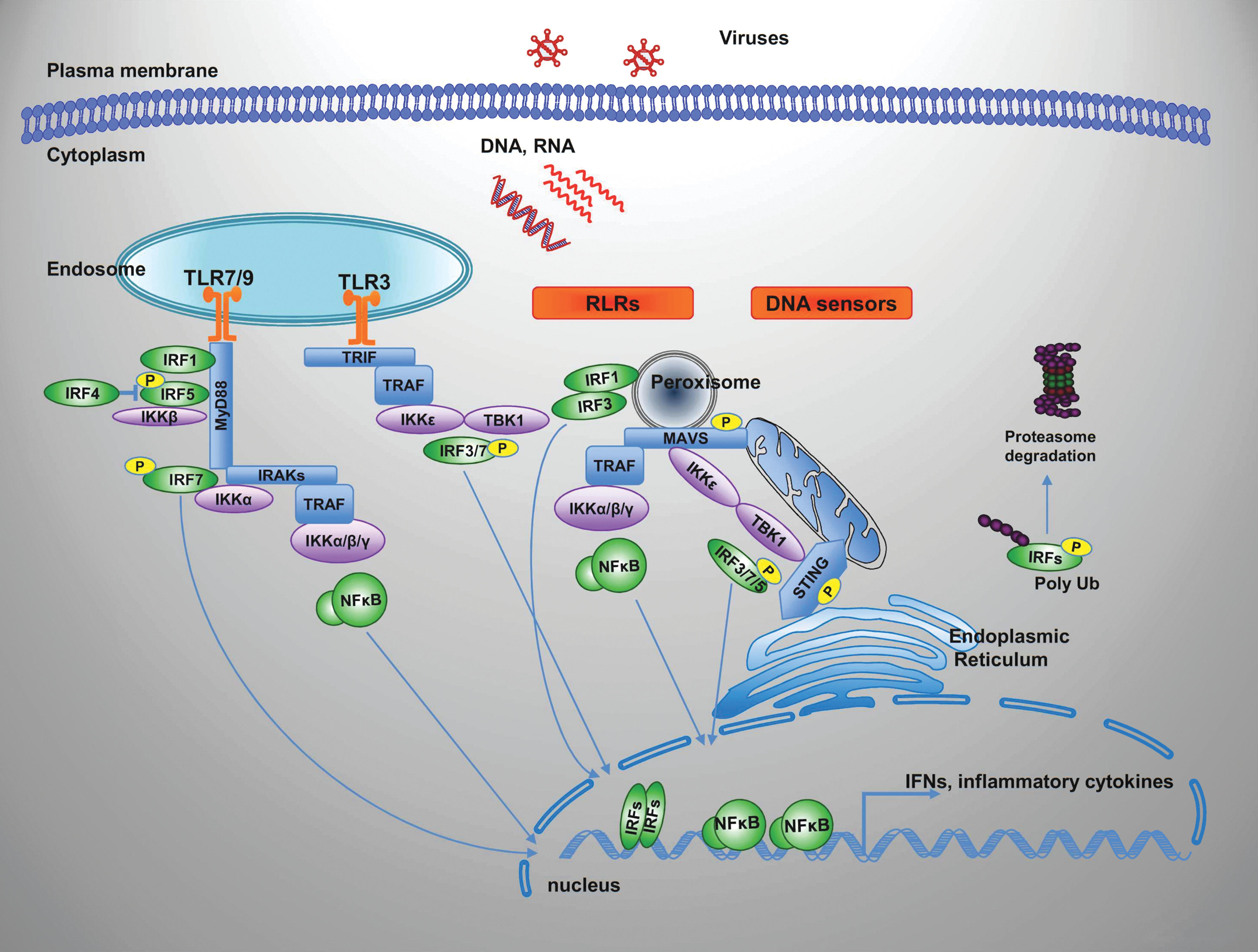
Summary of the pattern recognition receptor signaling pathways that recognize virus infection and activate IRFs. Innate sensing of viruses depends on detection of viral PAMPs, RNA or DNA by a range of cell surface and intracellular pattern recognition receptors, including TLR, RLR, and DNA sensors. These sensors signal through a limited number of shared adaptors and signaling molecules such as MYD88, TRIF, MAVS, and STING, initiating a cascade of events that lead to IRF and NF-κB activation from the IKK complex and the IKK-related kinases, TBK1/IKKɛ through TRAF family members. In specific cellular setting, as plasmacytoid dendritic cells, downstream TLR7/9, IRF7 binds to MyD88 and is activated by a protein kinase cascade that involves IKK-α. TRAF also interacts with and activates IRF7. IRF1 and IRF5 similarly interact and are activated by MyD88-dependent signaling. IRF4 binds to MyD88 in a region that overlaps the IRF5-binding region, thus inhibiting the binding of IRF5 to MyD88. Activated NF-κB and IRFs then translocate to the nucleus where they induce transcription of their specific target genes. See text for more details and references. IKK, inhibitor of κB kinase; NF-κB, nuclear factor-κB; PAMPs, pathogen-associated molecular patterns; RLR, RIGI-like receptor; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor. Color images available online at
The IRFs with intrinsic antiviral function include IRF3 and IRF7 that are essential for the PRR-mediated IFN gene transcription; IRF1 whose positive regulation of type I IFN gene expression occurs only in response to specific stimuli and in a cell-type- and time-specific manner (Negishi and others 2006; Dixit and others 2010; Hoshino and others 2010), but whose activation of type III IFN from peroxisomes is a universal mechanism independent of cell type or stimulus (Ueki and others 2013; Odendall and others 2014); and IRF9 that is integral to the type I and III IFN signaling pathways as part of the heterocomplex IFN-stimulated gene factor 3 (ISGF3) (Darnell and others 1994) that drives the expression of most ISGs. IRF1 and IRF7 are themselves under the control of IFN signaling for further amplification of the IFN response. In addition, IRF1, IRF3 and IRF7 have also intrinsic antiviral activities and the potential to induce ISGs in an IFN-independent manner. IRF5 is instead specifically involved in inflammatory cytokine induction (Takaoka and others 2005; Ryzhakov and others 2015) and is required for full induction of type I IFN genes in specialized cells as plasmacytoid dendritic cells (pDCs) downstream TLR7/9 signaling (Schoenemeyer and others 2005; Steinhagen and others 2013).
Given the unique function exerted by IRFs in mediating immune responses, it is not surprising that viruses have evolved a myriad of strategies aimed at rendering IRFs harmless. These strategies may involve a number of different molecular mechanisms, and more than 1 strategy may be necessary to overcome IRF activity by a single pathogen. Viruses may prevent or interfere with IRF activation indirectly by blocking PAMP recognition and activation of PRR signaling pathways or directly by (1) interfering with IRF activation and activity; (2) affecting IRF expression; and (3) hijacking and/or exploiting IRF activities.
Leaving out indirect strategies that envision IRF shutdown due to the viral ability to avoid detection in the first place and/or to interfere with sensor signaling pathways upstream IRF activation comprehensively reviewed elsewhere (Bowie and Unterholzner 2008; Versteeg and Garcia-Sastre 2010; Remoli and others 2011; Taylor and Mossman 2013; Zinzula and Tramontano 2013; Orzalli and Knipe 2014; Acchioni and others 2015; Coccia and Battistini 2015; Rosadini and Kagan 2015), here we summarize recent literature outlining evasion tricks utilized by some pathogenic viruses to directly disarm IRF.
Hit and Disarm
To disarm IRFs downstream PRR signaling pathways, viruses may block IRF activation and/or impede their transcriptional activity. Viral proteins may interfere and/or block IRF activation upon interaction/competition with IRF activators or with DNA target sequences and/or coactivators in transcription.
How viruses interfere with IRF activation
IRF activation and function are regulated by different and multiple PTMs where phosphorylation and ubiquitination/sumoylation have the more relevant impact in the antiviral response. These PTMs may be constitutive or induced by different stimuli.
IRFs have been shown to be phosphorylated at both serine/threonine and tyrosine residues. Phosphorylation is required for protein–protein interaction and formation of homo- and heterodimers with themselves and/or other transcription factors or cofactors and for binding to DNA target sequences and regulation of IRF transcriptional activity (Taniguchi and others 2001; Chen and Royer 2010; Battistini 2012).
Downstream PRR signaling, virus-induced phosphorylation is a major mechanism governing IRF3-, IRF5-, and IRF7-mediated induction of cytokines and chemokines. IRF phosphorylation, indeed, results in a conformational change that promotes dimerization, nuclear localization, association with the coactivator CBP/p300, and binding to target sequences to stimulate target promoter activity (Kumar and others 2000; Lin and others 2000; Servant and others 2002; Qin and others 2003; Chen and others 2008b).
In most cell types, 2 atypical IKK-related kinases, the inhibitor of κB kinase (IKK)-ɛ and the TANK-binding kinase 1 (TBK1), are responsible for phosphorylation of IRF3 and IRF7 at specific serine/threonine residues in the C-terminal portion of these proteins (Fitzgerald and others 2003; Sharma and others 2003). In the specific setting of TLR7/9-mediated dendritic cell (DC) activation, IRF7 phosphorylation is instead mediated by IKKα (Hoshino and others 2006). In the same setting, IRF5 (Lopez-Pelaez and others 2014) and IRF-1 (Hoshino and others 2010) can also be phosphorylated by IKKα. In addition, 2 recent independent studies (Lopez-Pelaez and others 2014; Ren and others 2014) provided evidence that IRF5 is also phosphorylated at a single site at serine 446 by IKKβ, a component of the IKK complex specifically involved in NF-κB activation (Hayden and Ghosh 2012), leading to coordinated stimulation of proinflammatory genes.
Since phosphorylation is absolutely required for IRF activity in response to pathogen detection, it is not surprising that IRF-activating kinases are specific targets of viral evasion strategies. These strategies include direct interaction of viral proteins with the kinase or with components of the kinase-activating complexes that can result in the kidnapping of the kinase, competition with the physiological substrate, and/or in the inhibition of the catalytic kinase activity. Examples are provided of such viral strategies (Fig. 3A) below.
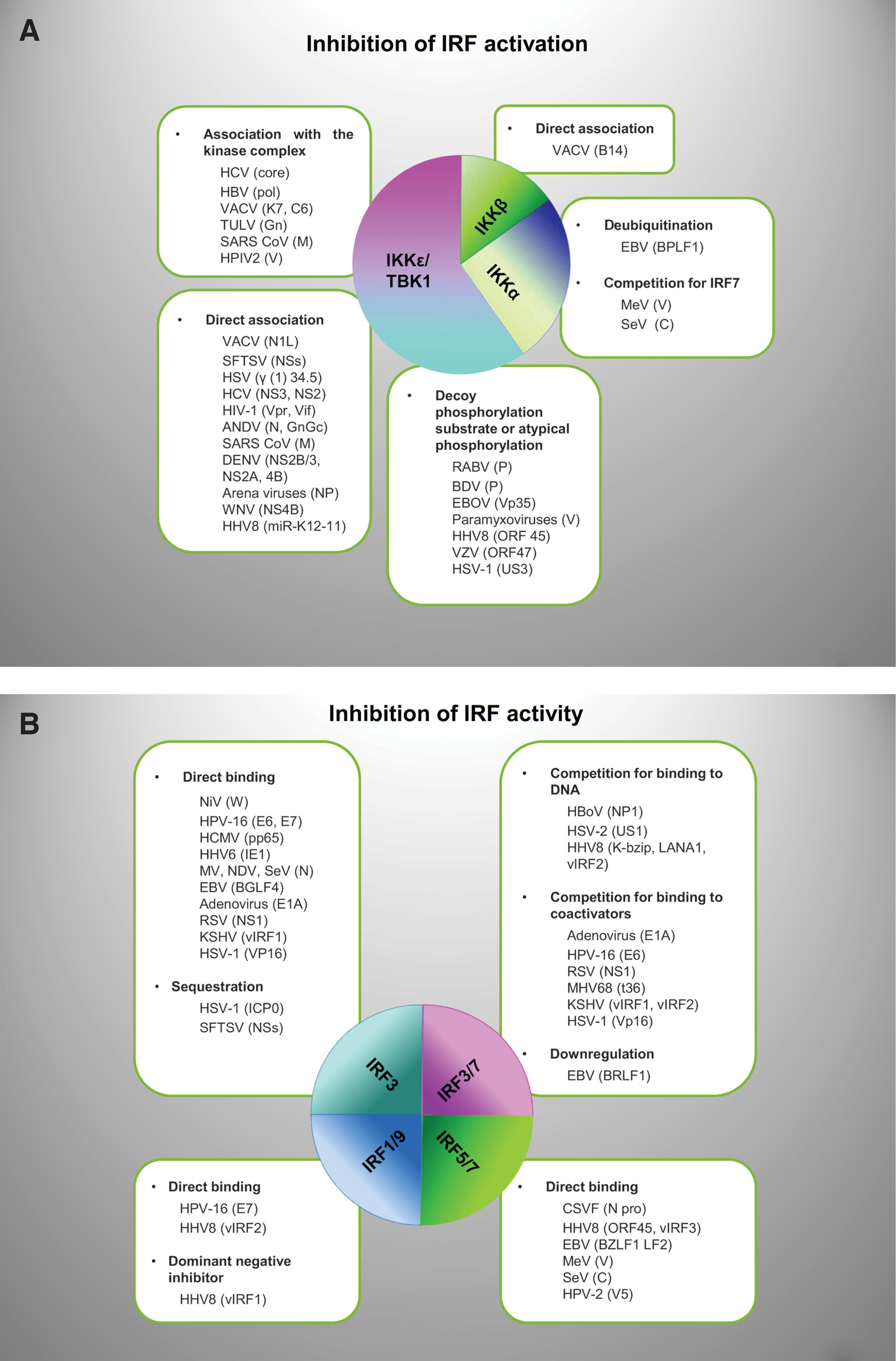
Examples of viral antagonists that block, subvert, or exploit IRFs by multiple mechanisms. Virus proteins may affect both activation and activity of IRFs
The N1L from vaccinia virus (VACV) blocks IRF3/7 activation by associating with several components of the multisubunit I-kappaB kinase complex, most strongly with TBK1 (DiPerna and others 2004). The NSs protein from the novel Severe Fever with Thrombocytopenia Syndrome Phlebovirus (SFTS) in the Bunyaviridae family and the gamma(1)34.5 protein of herpes simplex viruses (HSVs) (Verpooten, Ma, and others 2009, Ma, Jin, and others 2012, Qu, Qi, and others 2012) and the hepatitis C virus (HCV) serine protease NS3 (Otsuka and others 2005) were similarly found to associate preferentially with TBK1 and to compete for the binding to IRF3, thus dampening IRF3 activation. Moreover, it has been recently reported that in infected cells, NSs of SFTS virus upon interaction with TBK1 form viral inclusion bodies in which they sequester the IKK complex consisting of TBK1, IKK-ɛ, and even activated IRF3 (Wu and others 2014).
The NS2B/3 serine protease of dengue virus (DENV), instead, directly interacts with IKK-ɛ, masking the kinase domain and blocking phosphorylation and nuclear translocation of IRF3 (Anglero-Rodriguez and others 2014). Moreover, the NS2A and NS4B from DENV serotypes 1/2/4, as well as the West Nile virus (WNV) NS4B protein dose dependently inhibit the phosphorylation of TBK1 and IRF3, suggesting that they function at the level of TBK1 complex activation. A comparative analysis of NS4A proteins across DENVs demonstrated that DENV1, but not DENV2 or DENV4, NS4A proteins uniquely inhibited TBK1, suggesting that NS4A is a specific virulence factor that contributes to the increased severity of DENV1 infections compared with other less pathogenic serotypes (Dalrymple and others 2015).
The NS2 protein of HCV inhibits, in a dose-dependent manner, both IKK-ɛ- and TBK1-induced IRF3 phosphorylation by directly interacting with the kinases (Kaukinen and others 2013). HCV core, instead, inhibits IRF3 dimerization as well as phosphorylation induced by Newcastle disease virus (NDV) infection or poly (I:C), through the basic amino acid region 1 (BR1) in the NH2-terminal region of the protein (Inoue and others 2012). This domain is the binding region for a DEAD box protein, the DDX3, a helicase, interaction partner of TBK1 and IKK-ɛ kinases that are required to enhance the TBK1/IKK-ɛ-induced IFN-β promoter activity upon binding to the adaptor MAVS (Soulat, Burckstummer and others 2008; Oshiumi and others 2010). The HCV core also decreases the expression levels of DDX3, further reinforcing the inhibition of IRF3 activation. Likewise, the hepatitis B virus (HBV) polymerase and the K7 protein from VACV (Schroder and others 2008) function dampening the interaction between TBK1/IKK-ɛ and DDX3 (Yu and others 2010). Another virulence factor of VACV, the B14 protein, instead has been shown to inhibit NF-κB activation by interacting with the IKKβ component of the IKK complex (Chen and others 2008a). Since, as mentioned above, IKK-β also phosphorylates IRF5, it is tempting to speculate that by targeting this kinase, VACV B14 inhibits also IRF5. Whether this occurs in the context of viral infection and in which cell setting IRF5 inhibition is physiologically relevant, however, remain to be tested. In contrast, the VACV protein C6 inhibits IRF3/7 activation and translocation into the nucleus, but does not inhibit NF-κB activation. C6, as K7, acts downstream of TBK1 and IKK-ɛ by interacting with the kinase scaffold adaptor proteins, TANK, SINTBAD, and NAP1 (Unterholzner and others 2011). Inhibition of TBK1 activity through binding to the TBK1 complex component, TRAF3, is instead exerted by the Tula virus (TULV) Gn protein (Matthys and others 2011). Similarly, the Severe acute respiratory syndrome-Corona virus (SARS-CoV) M protein physically associates with RIG-I, TBK1, IKK-ɛ, and TRAF3, preventing the formation of functional complex activating kinases (Siu and others 2009).
The nucleocapsid protein NP proteins from several Arenaviruses associate with the kinase domain of IKK-ɛ, but not TBK1, and sequester it in a complex that prevents IKK-ɛ binding to the adaptor MAVS, thus blocking its autocatalytic activity and preventing IRF3 phosphorylation (Pythoud and others 2012).
Autophosphorylation of TBK1 is also inhibited by human immunodeficiency virus-1 (HIV-1) Vpr and Vif upon binding to TBK1 (Harman and others 2015). This occurs in myeloid cells that represent the portal of virus entry. Interestingly, in T cells, the same viral proteins instead block IFN induction by targeting IRF3 for cellular degradation, highlighting the fine-tuned mechanisms utilized by HIV-1 to interfere with innate recognition by exploiting multifunctional accessory proteins, as better discussed below.
Interference with TBK1 activation has been reported also for the nucleocapsid (N) proteins of Hantaviruses. The N protein of the Andes virus (ANDV) that is distinguished from other hantaviruses by its unique ability to spread from person to person and to cause highly lethal hantavirus pulmonary syndrome in patients, in contrast to other hantavirus N proteins, inhibits TBK1-directed IRF3 phosphorylation at serine 396 and TBK1 autophosphorylation at serine 172 (Cimica and others 2014). Similarly, ANDV GnGc protein blocks TBK1-directed IRF3 phosphorylation, but at a downstream step since it does not inhibit TBK1 autophosphorylation.
The phosphoproteins of many nonsegmented negative-strand RNA viruses, such as rabies virus (RABV), Borna disease virus (BDV), and ebola virus (EBOV), also interact with IRF-activating kinases functioning as alternative substrates and preventing activation of the physiological substrate. This is the case of the rotavirus (RV) phosphoprotein P (Brzozka and others 2005), the Vp35 from EBOV (Prins and others 2009), and the P protein of BDV (Unterstab and others 2005). By interacting with the TBK1/IKK-ɛ kinase domain, these proteins function as a substitute substrate, inhibiting both the kinase activity and the binding of the physiological IRF3/7 substrate. Moreover, the Vp35 and the P protein from BDV are phosphorylated themselves by TBK1.
The V proteins of several paramyxoviruses, including mumps virus (MuV) and human parainfluenza virus (HPIV) 2 and 5, also serve as a decoy substrate for TBK1/IKK-ɛ by mimicking IRF3 and thus competing with cellular IRF3 for phosphorylation (Lu and others 2008).
Two viral kinases, the ORF47 of varicella-zoster virus (VZV), an alphaherpesvirus restricted to humans, and the Us3 protein of HSV-1, instead, phosphorylate IRF3 in an atypical and TBK1-independent manner, thus preventing proper activation by host kinases (Vandevenne and others 2011; Wang 2013 #62; Wang and others 2013). Interestingly, another protein of VZV, the viral immediate-early protein 62 (IE62), blocks TBK1-mediated IFN-β secretion and IRF3 function by directly blocking phosphorylation of at least 3 serine residues on IRF3. However, the exact mechanism has not been elucidated since IE62 binding to TBK1 or IRF3 was not detected and it was shown that IE62 does not perturb TBK1-IRF3 complex formation (Sen and others 2010).
The β-herpesvirus human cytomegalovirus (HCMV) also encodes a protein, pp65, which subverts IRF3 activity by inhibiting its nuclear accumulation probably due to decreased phosphorylation (Abate and others 2004). The IE protein 5 of the related β-herpesvirus, HHV-6, similarly inhibits the nuclear localization of IRF3, leading to a decreased binding to the IFN-β gene promoter by decreasing IRF3 phosphorylation (Jaworska and others 2007).
At variance with IRF3, IRF7 is expressed at very low levels in most cell types and induced by IFN-mediated signaling, while it is expressed at discrete levels only in specialized immune cells, which potently induce type I IFN expression. Nevertheless, even small amounts of IRF7 are crucial in most cell types even in the initial phase of IFN gene induction by virus-engaged PRRs (Sato and others 2000). Moreover, IRF7 has been reported to confer an ISGF3-like transcriptome signature onto the cell (Schmid and others 2010), allowing the early establishment of a hostile environment for viral replication. Thus, IRF7 is a hit target of virus-mediated inhibitory mechanisms that not only mirror those utilized to disarm IRF3 along activation pathways shared by the 2 IRFs, as described above, but that may also target unique activation of IRF7.
The ORF45 of the human herpes virus 8 (HHV8), also known as Kaposi's sarcoma-associated herpesvirus (KSHV), serves as an alternative substrate for TBK1/IKK-ɛ and competes with IRF7, but not IRF3, for phosphorylation by these kinases, implying that ORF45 does not inactivate the kinases directly. Consistently, as the phosphoproteins of many nonsegmented negative-strand RNA viruses described above, ORF45 is also phosphorylated efficiently on Ser41 and Ser162 by IKK-ɛ and TBK1 (Liang and others 2012).
Interestingly, KSHV also encodes miRNA, the miR-K12-11, which directly targets and downregulates IKK-ɛ mRNA translation, reinforcing its ability to impair, in this case, both IRF3 and 7 phosphorylation. Moreover, since inhibition of miR-K12-11 enhanced KSHV reactivation induced by vesicular stomatitis virus (VSV) infection, it can be speculated that the targeting of IKK-ɛ contributes to maintenance of KSHV latency (Liang and others 2011).
Competition with IKKα is another mechanism to specifically block IRF7. The V protein of measles virus (MeV) abolishes TLR7/9-dependent IFN induction in human plasmacytoid DCs by competing with IRF7 for binding to the kinase IKKα. Moreover, the binding of V protein to IKKα results in phosphorylation of the viral protein at the expense of IRF7 phosphorylation both in vitro and in living cells. The V protein also binds to IRF7, directly inhibiting IRF7 polyubiquitination-mediated activation and transcriptional activity (Pfaller and Conzelmann 2008). Likewise, in pDCs, the V protein from human parainfluenza virus type 2 not only binds to IKKα, preventing IRF7 activation, but also interacts with TRAF6, quenching IRF7 transcriptional activity by inhibiting TRAF6-mediated Lys63 polyubiquitination of IRF7 (Kitagawa and others 2011, 2013), a prerequisite for its activation (Ning and others 2008). Interestingly, the Trp-rich motif present in the C-terminus of the V protein and mediating the inactivation of IRF7 is distinct from that required for other evasion strategies exerted by paramyxovirus V proteins (Nishio and others 2002; Ramachandran and Horvath 2010).
IKKα-mediated IRF7 phosphorylation, downstream TLR7/9 in pDC, is also blocked by the interaction of IKKα with the C protein of Sendai virus (SeV), another paramyxovirus (Yamaguchi and others 2014). Interestingly, this anti-IFN activity of the C protein is shared across the 3 genera of the Paramyxovirinae Respirovirus, Morbillivirus, and Henipavirus supporting an important role for IFN production by pDC in the host response to paramyxovirus infection.
How viruses interfere with IRF activity
Through their IAD motif, IRFs can associate with other IRFs or other transcription factors and coactivators of transcription to form complexes that modulate their ability to bind DNA target sequences and regulate gene transcription (Taniguchi and others 2001). A block of IRF activity by viruses and their genetic products may thus be achieved by direct binding to IRFs, thus inhibiting IRF association with other regulators and/or association with DNA target sequences; by competition with IRF for the binding to DNA or to coactivators, and by recruitment and sequestering of IRF to cell structures physically far from active chromatin.
The following are examples of each of these strategies (Fig. 3B).
Direct binding of viral proteins to IRF has been recognized early on. The E6 protein from the oncogenic human papillomavirus type 16 (HPV-16) directly binds IRF3 and inhibits its transactivation potential (Ronco and others 1998). The ISGF3 complex is also dismantled by the HPV-16 E7 oncoprotein by direct protein–protein interaction with and inhibition of nuclear translocation of IRF9 (Barnard and McMillan 1999). The HCMV instead decreases the expression of IRF9, similarly impeding the formation of the ISGF3 complex (Miller and others 1999). Nuclear translocation by direct binding to IRF3 is impeded by the cytoplasmic V protein of several paramyxoviruses, including MeV, SeV, and NDV, but not by the V proteins of MeV and Nipah virus (NiV) (Irie and others 2012). NiV uses the W protein to inhibit IRF3 activity by binding and sequestering an inactive IRF3 in the nucleus where NiV W localizes, thus preventing interaction with the cytoplasmic activating kinases TBK1/IKKɛ (Shaw and others 2005).
The Npro of classical swine fever virus (CSFV) interacts with IRF7 through its Zn-binding domain and this results in lower protein levels and impairment of IRF7 activity in pDCs (Fiebach and others 2011).
The viral immediate-early protein, ORF45 of HHV8, in addition to serving as an alternative substrate for kinases, as described above, also specifically associates with IRF7 inhibitory domain stabilizing an IRF7 autoinhibitory conformation. This further reinforces inhibition of IRF7 phosphorylation and blocks its translocation to the nucleus (Sathish and others 2011). HHV8 also encodes 4 viral IRF (vIRF-1–4) analogs that directly interfere with host IRF activity (Jacobs and Damania 2011; Lee and others 2012). In particular, vIRF3 interacts with and inhibits the DNA binding activity of IRF5 and IRF7 (Joo and others 2007; Wies and others 2009). vIRF3 specifically interacts with either the DNA binding domain or the central IRF association domain of IRF7.
The Epstein-Barr virus (EBV) immediate-early protein BZLF-1 binds IRF7 and, even if this had no effect on the nuclear localization of IRF7, prevents it from activating type I IFN promoters (Hahn and others 2005; Ning and others 2011). Another EBV product, the viral tegument protein, LF2, binds the IRF association domain of IRF7 and prevents its dimerization (Wu and others 2009). The EBV kinase, BGLF4, instead interacts with IRF3 and despite the fact that it does not alter either IRF3 dimerization or nuclear translocation and CBP recruitment, it prevents IRF3 from binding to responsive promoters (Wang and others 2009).
So far, only few viruses have been shown to compete with activated IRF for the binding to target promoters. The NP1 protein of the human bocavirus (HBoV) and the immediate-early protein of HSV-2 US1 directly interact with the DNA binding domain of IRF3, preventing its association with the IFN-β gene promoter (Zhang and others 2012, 2015). Similarly, 2 proteins of KSHV, the latency-associated nuclear antigen (LANA-1) (Cloutier and Flamand 2010) and the K-bZIP leucine zipper-containing transcription factor encoded by ORFK8 (Lefort and others 2007), interfere with the binding of IRF3 to the IFN-β promoter. Interestingly, the K-bZIP binds to the IFN-β gene promoter, preventing IRF3 attachment and the formation of the enhanceosome. This results in the triggering of low-level activation of the IFN-β gene, but in the prevention of proper robust activation by physiological inducers. On the other hand, by competing with IRF3 for the binding to DNA target sequences, the K-bZIP also blocks the stimulation of primary IRF3 target genes.
Another strategy to block IRF activity is to compete for and/or inhibit the binding to coactivators such as CBP and p300 preventing the formation of an enhanceosome and the recruitment of RNA polymerase II for gene transcription activation (Agalioti and others 2000).
This is the case of the adenovirus (AdV) E1A gene product (Juang and others 1998) and of the E6 protein from HPV-16. The E1A not only directly binds IRF3 but also competes for the IRF3 binding to CBP/p300. Similarly, HPV-16 E6, in addition to direct IRF3 interaction, also binds to 3 regions (C/H1, C/H3, and the C-terminus) of both CBP and p300 (Patel and others 1999) competing for the binding to IRF3.
Likewise, the respiratory syncytial virus (RSV) NS1 protein associates with both IRF3 and CBP, preventing their interaction (Ren and others 2011). Interaction between IRF3 and CBP is also impeded by a conserved herpesviral kinase, designated t36 in murine γ-herpesvirus 68 (MHV-68), which specifically binds to the activated form of IRF3 in the nucleus (Hwang and others 2009). The ORF K9 of HHV8, also known as vIRF1, the homolog of IRF1, binds to both IRF3 and IRF7, but it blocks only IRF3-mediated transactivation by preventing IRF3 recruitment of the CBP/p300 coactivators (Burysek and others 1999; Lin and others 2001). vIRF2 inhibits the expression of ISGs driven by IRF1 and IRF3, but not IRF7, but with a mechanism apparently divergent from that of vIRF1 (Fuld and others 2006). In addition, vIRF2 targets STAT1 and IRF9 components of ISGF3 complex also impairing the IFN signal transduction pathway (Mutocheluh and others 2011).
HSV-1 uses 2 proteins, ICP0 and VP16, to inhibit IRF3 activity. ICP0 recruits activated IRF3 and CBP/p300 to nuclear structures away from the host chromatin and also leads to the inactivation and accelerated degradation of IRF3 (Melroe and others 2007), as better described below. ICP0 also inhibits IRF7-mediated induction of ISGs, but it remains to be established whether the same mechanism of IRF3 inactivation is involved (Lin and others 2004). This effect seems, however, relevant in vivo since pDCs that endogenously express high levels of IRF7 play a key role during the early response to HSV-1 infection (Rasmussen and others 2007). VP16 instead binds to IRF3, but not IRF7, in vivo.
As ICP0, VP16 does not affect IRF3 dimerization, nuclear translocation, or DNA binding activity, but competes for the IRF3 binding to CBP, efficiently inhibiting the formation of the transcriptional complexes IRF3-CBP (Xing and others 2013). Interestingly, CBP is also a common target of distantly related human herpes viruses-encoded miRNAs (Cox and others 2015). This finding underscores the importance of this strategy to combat a host defense by distantly related viruses that results in evading both the induction and effector phases of the IFN response being CBP involved in the expression of both IFN synthesis and signal transduction. On the other hand, in HSV infection, CBP suppression may also promote latency by restricting lytic activation (Du and others 2013). The role of IRF targeting in virus-induced persistent/latent infection is described in a following section.
Hit and Deactivate or Destroy
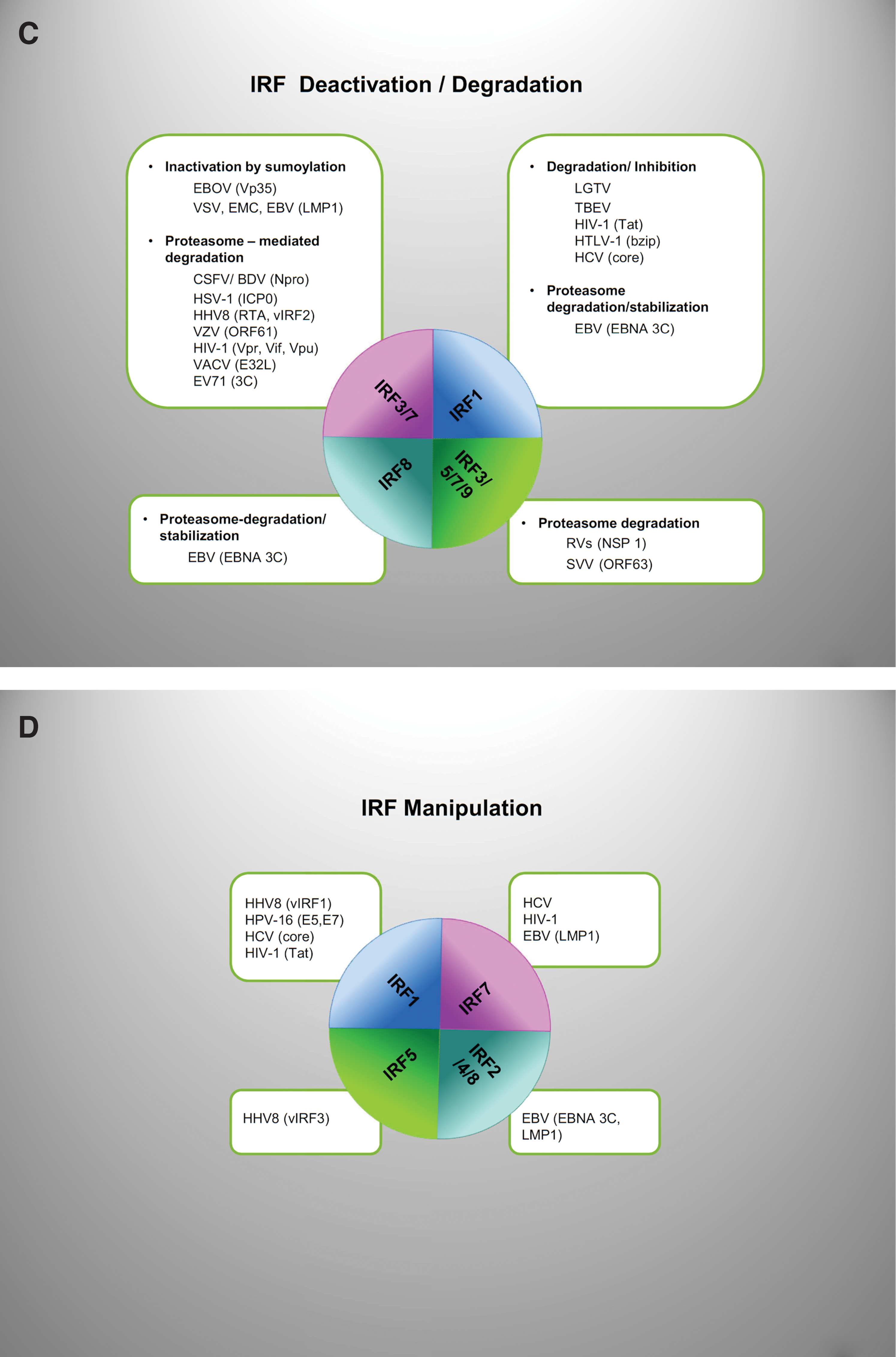
Viruses have evolved strategies that impede IRF functions also through IRF deactivation or modulation of IRF expression and stability (Fig. 3C). These strategies are largely dependent on the viral ability to exploit the ubiquitin system.
Over the past few years, mounting evidence has demonstrated the essential roles of ubiquitination in serving as an effective, rapid and versatile means to trigger defense mechanisms against RNA or DNA viruses, as well as to prevent premature or excessive activation of PRR-induced signaling (Davis and Gack 2015; Liu and others 2015). As such, a number of viruses target host proteins, including IRFs, exploiting the ubiquitin system to deactivate and/or degrade them (Viswanathan and others 2010; Gustin and others 2011). The cellular fate of ubiquitinated proteins varies greatly depending on the type of conjugated ubiquitin chains linked on the modified residue of the substrate (Ikeda and others 2010; Komander and Rape 2012). Specifically, K63-linked polyubiquitination modulates nonproteolytic processes, including membrane trafficking and signal transduction, whereas K48-linked polyubiquitination generally targets the substrates for proteasomal degradation.
A number of adaptor signaling proteins in the PRR cascade, including members of the IRF family, are activated by K63-linked ubiquitination. On the other hand, ubiquitin-mediated degradation of IRFs is physiologically used as an effective mechanism to dampen IFN overexpression and/or to regulate IRF stability in physiological conditions (Higgs and Jefferies 2008; Battistini 2012; Davis and Gack 2015; Liu and others 2015). Thus, the multifaceted nature of the ubiquitination pathway for protein regulation makes it an attractive target of many viruses.
Viruses may encode E3 ligases or deubiquitinases or may redirect the cellular E3 ligases and deubiquitinases to modulate the ubiquitination status of substrates of choice (Randow and Lehner 2009). By using viral proteins that can function as deubiquitinases, viruses may block the innate immune system by removing ubiquitin from the several components of the RLR that are activated by ubiquitination and, in a specular manner, may employ viral or host ubiquitin ligases to target immunoproteins for proteasomal or lysosomal degradation through K48-linked ubiquitination.
Despite that several of these viral components have been shown to exploit the ubiquitin system to their own advantage, by targeting and inactivating especially components of the pathway upstream IRFs (Viswanathan and others 2010; Gustin and others 2011; Jiang and Chen 2012), some examples of direct ubiquitination/deubiquitination of IRFs by viral proteins have been also described and few of them are reported below.
As an ubiquitin homolog, small ubiquitin-related modifiers (SUMOs) cause the sumoylation of IRF3 and IRF7. In DCs, EBOV VP35, by interacting with the small ubiquitin-like modifier SUMO E2 enzyme Ubc9 and the E3 ligase PIAS1, promotes IRF7 sumoylation, a PTM that prevents IRF translocation into the nucleus (Chang and others 2009). In this case, the virus exploits a physiological pathway. SUMO modification of IRF3/7, negatively regulating their transcriptional activity, is indeed part of the negative feedback loop that contributes to postactivation attenuation of IFN production (Kubota and others 2008).
IRF3 and IRF7 SUMOylation is also exploited by VSV and encephalomyocarditis virus (EMCV). Upon VSV infection, IRF3 and IRF7 are modified by SUMO1, SUMO2, and SUMO3 (Kubota and others 2008). Interestingly, SUMOylation of IRF3 and IRF7 was not dependent on their phosphorylation, and vice versa (Kubota and others 2008).
EBV latent membrane protein 1 (LMP1) regulates an intricate mechanism that affects the equilibrium between lytic replication and maintenance of latency, as better described below. By inducing TRAF6-mediated ubiquitination and activation of IRF7, it can induce lytic replication (Ning and others 2008), but on the other hand, LMP1 also induces the cellular deubiquitinating enzyme A20, which inactivates IRF7 through deubiquitination (Ning and Pagano 2010). Furthermore, in latently infected cells, LMP1 inactivates IRF7 also by sumoylation (Bentz and others 2012). Sumoylation regulates the expression and function of IRF7 by decreasing its turnover, increasing its nuclear retention, decreasing its DNA binding, and limiting its transcriptional activation. IRF7 inactivation has thus the double advantage to inhibit the early immune response to avoid virus clearance, while favoring establishment and maintenance of latency, as better described below.
Another EBV protein, BPLF1, is endowed with deubiquitinase activity and blocks IRF7 activity indirectly by deubiquitinating IKKα (van Gent and others 2014).
As mentioned above, targeting for proteasomal degradation, upon K48-linked polyubiquitination, is a widely reported mechanism not only to modulate protein stability in homeostatic conditions but also exploited by viruses to get rid of cellular proteins that may interfere with their survival.
Proteasome-dependent ubiquitin-mediated degradation of IRF3 is used by flaviviruses as the pestivirus CSFV and the bovine viral diarrhea virus (BVDV). The Npros of both viruses interact with IRF3, before its activation by phosphorylation, and mediate its polyubiquitination and subsequent proteasomal degradation. This allows the viruses to establish a productive infection in their primary target cells, such as myeloid DCs and endothelial and epithelial cells (Bauhofer and others 2007; Chen and others 2007; Seago and others 2007).
Accelerated proteasome-dependent degradation of IRF7 in most cell type except lymphoid tissue infected with NDV has also been reported (Prakash and Levy 2006).
Several herpesviruses also actively induce K48-linked ubiquitination and proteolysis of IRF3 and 7 using their immediate-early protein ICP0, which exhibits E3 ligase activity. However, the mechanism of viral evasion mediated by ICP0 appears to be only partially conserved between herpesviruses. Indeed, in contrast to HSV-1-encoded ICP0 that (as described above) interacts with IRF3 sequestering it and its coactivator CBP/p300 in nuclear bodies (Melroe and others 2007), ICP0 encoded by bovine herpesvirus 1 reduces IRF3, but not IRF7, levels, directly or indirectly targeting IRF3 for proteasome-dependent degradation (Saira and others 2007).
The ORF50 (RTA) of HHV8, acting as an E3 ubiquitin ligase, promotes IRF7 ubiquitination and the proteasome-mediated degradation of IRF7 (Yu and others 2005). In addition, it was shown that RTA can recruit the cellular E3 ubiquitin ligase RAUL to IRFs and cooperate with RAUL to catalyze K48-linked polyubiquitination of both IRF3 and IRF7, inducing their proteasome-mediated degradation (Yu and Hayward 2010). RAUL is, indeed, the ligase that physiologically leads to both IRF7 and IRF3 degradation to negatively regulate IFN expression.
VZV immediate-early protein ORF61 directly interacts with activated IRF3 and uses its RING finger E3 ubiquitin ligase domain to ubiquitinate and degrade IRF3 through the proteasome (Zhu and others 2011). IRF9 is similarly degraded in a proteasome-dependent manner by the simian varicella virus (SVV) ORF63 (Verweij and others 2015).
In T cells, the HIV-1 virion-associated accessory proteins, Vif, Vpr, and Vpu, acting as E3 Ub adaptor proteins, mediate ubiquitination of IRF3 and target IRF3 for proteasome-mediated degradation (Okumura and others 2008; Doehle and others 2009, 2012). Vpu-mediated IRF3 degradation is, however, still debated. Recently, it has indeed been reported that actually Vpu effect on IRF3 involves a partial cleavage of IRF3 in a caspase-dependent manner. This results in a terminal fragment that can act as a negative regulator of IRF3-dependent gene activation and induced apoptosis (Park and others 2014). Likewise, the HHV8 vIRF2 accelerates IRF3 degradation by a mechanism exploiting caspase-3 that (as RAUL) participates in normal IRF3 turnover (Areste and others 2009).
The RV nonstructural protein NSP1, acting as a putative E3 ubiquitin ligase, induces, with similar efficiencies, the proteasome-dependent degradation of not only IRF3 (Graff and others 2007) but also of IRF5 and IRF7 (Barro and Patton 2007). This multiple attack could allow the virus to cross the gut barrier by replicating in specialized trafficking cells (eg, macrophages and DCs) that constitutively express IRF7. Interestingly, human RVs rely predominantly on the NSP1-induced degradation of IRF5 and IRF7, while animal RVs target IRF3, IRF5, and IRF7, allowing the animal viruses a broader attack on the IFN signaling pathway (Arnold and Patton 2011). Recently, it has been reported that IRF9, but not IRF1, is similarly targeted for degradation by NSP1. Interestingly, NSP1 requires a structurally intact IAD1 for recognition and targeting of IRF proteins, and consistently IRF1, which lacks a classical IAD is not degraded (Arnold and others 2013).
An indirect effect on IRF3 stability is instead exerted by the E3L protein of VACV that targets and disables ISG15 (Guerra and others 2008). ISG15 is an ISG that besides other functions covalently binds IRF3 and stabilizes it by preventing its ubiquitin-mediated degradation in NDV-infected cells (Lu and others 2006).
Specific cleavage of IRF7, but not of IRF3, is exerted by the 3C protein from the enterovirus 71 (EV71) through a process insensitive to inhibitors of caspase, proteasome, lysosome, and autophagy. 3C protein is indeed endowed with protease activity and directly mediates cleavage of IRF7 within the constitutive activation domain, resulting in 2 cleaved fragments that are no more able of activating IFN expression. On the other hand, expression of the amino-terminal domain of IRF7 enhances viral infection consistent with its ability to interact with and inhibit IRF3 (Lei and others 2013).
Finally, downregulation of IRF3 and IRF7 transcription is exerted by the EBV-encoded protein BRLF1 during the lytic cycle (Bentz and others 2010).
As IRF3 and IRF7, IRF1 is regulated by ubiquitin-mediated proteosomal degradation controlled by the carboxyl-terminal domain of the protein (Nakagawa and Yokosawa 2000; Pion and others 2009). IRF1 is the most versatile member of the IRF family, implicated in a variety of cellular functions spanning from the development and function of various immune cells to antiviral and tumor suppression activity. Despite being identified as a regulator of type I IFN gene expression, IRF1 is actually considered not essential for IFN gene expression except in specific settings and cell types, making the IRF1-mediated IFN-β induction a tailored response (Negishi and others 2006; Yarilina and others 2008; Venkatesh and others 2013). Nevertheless, IRF1 is involved in IFN-induced antiviral and antibacterial immunity and recently its intrinsic antiviral activity and its direct role in controlling IFN-independent signaling events have been also defined (Schoggins and others 2011; Schoggins 2014).
IRF1 also plays a crucial role in regulating peroxisomal MAVS-dependent signaling from peroxisomes inducing a rapid IFN-independent expression of antiviral factors that provide a rapid and short-term protection upstream the mitochondrial MAVS IRF3-activated IFN (Dixit and others 2010). Moreover, IRF1 is not dispensable for peroxisomal selective induction of type III IFN expression in viral-infected epithelial cells even independently from type I IFN expression (Odendall and others 2014). IRF-1 may thus provide an antiviral defense particularly important for those pathogens that evade innate immunity by disrupting the induction and function of IFN in the first place. Targeting of IRF1 by viruses is mainly exerted at the level of protein stability and protein expression.
Two tick-borne flaviviruses, the Langat virus (LGTV) and the tick-borne encephalitis virus (TBEV), have been recently reported to inhibit IRF1 independently of their ability to antagonize IFN signaling. In DCs infected with LGTV, a naturally attenuated member of the TBEV serogroup, the repression of interleukin (IL)-12 expression and DC maturation is due to diminished IRF1 protein levels. Similar effects are observed with the highly virulent Sofjin strain of TBEV (Robertson and others 2014).
In HIV-1-infected cells, Tat exploits the cellular E3 ligase HDM2 to affect IRF1 protein stability favoring K48-linked polyubiquitination of IRF1, preventing its antiviral and immunomodulatory activities to favor virus replication (Remoli AL, Battistini A, unpublished).
The HTLV-1 bZIP factor that is constitutively expressed in adult T-cell leukemia cells interacts with IRF1, reducing IRF1 stability through a proteasome-dependent pathway. IRF1-mediated apoptosis is also significantly reduced by ectopic production of the bZIP factor, suggesting that suppressive effects on IRF1 function may contribute to HTLV-1-induced cellular leukemogenesis (Mukai and Ohshima 2011).
IRF1 expression is, instead, suppressed at the transcriptional level by the HCV core in hepatic cells expressing the entire HCV replicon. This event blocks the expression not only of several antiviral genes but also of immunomodulatory genes involved in adaptive immunity and inflammation. In doing so, HCV core facilitates the establishment of HCV persistent infection (Ciccaglione and others 2007) as better described below.
Hijack and Manipulate or Exploit
Hijacking and harnessing or exploiting host pathways and proteins is a common strategy utilized by viruses to facilitate not only viral replication but also persistence and/or virus-induced carcinogenesis; these effects being only in part dependent on interference with the host IFN response (Li and others 2014; Acchioni and others 2015; Snell and Brooks 2015; Zitvogel and others 2015). Most IRFs are pleiotropic factors whose functions are not limited to activation and regulation of antiviral innate immunity, thus they are also directly manipulated and exploited by viruses to favor persistence and/or virus-induced carcinogenesis.
Examples of viruses that exploit IRFs to such purposes include KSHV, HPV, HCV, HIV-1, and EBV; IRF1 and IRF7 being the most widely targeted IRFs (Fig. 3D).
The human HHV8 has developed unique mechanisms for altering cellular proliferative and apoptotic control pathways by incorporating into its genome viral homolog to several cellular regulatory genes, including 4 viral interferon regulatory factors (vIRF1–4). These analogs, as mentioned above, act as dominant-negative factors that directly interfere with host IRFs (Jacobs and Damania 2011; Lee and others 2012).
The vIRFs are all expressed during lytic reactivation, but vIRF3 has also been detected in latently infected cells. In particular, vIRF3 is expressed in latently HHV8-infected primary effusion lymphoma cells and is essential for the survival of these cells. Moreover, as reported above, vIRF3 binds IRF5 and the importance of vIRF3/IRF5 interaction is not limited to inhibition of the IFN system. Indeed, IRF5-mediated cell growth regulation is significantly altered by overexpression of vIRF3 in B cells and vIRF3, by releasing IRF5 from p21 promoter transcription complexes, prevents IRF5-mediated growth inhibition and G2/M cell cycle arrest (Bi and others 2011). Similarly, by acting as dominant-negative inhibitors of IRF1, vIRF1 may inhibit IRF1 oncosuppressor functions (Takaoka and others 2008; Savitsky and others 2010; Fragale and Battistini 2013; Dou and others 2014). Recently, it has been reported that v-IRF4 that is expressed in the lytic phase, suppresses expression of c-IRF4 and, by competing with c-IRF4 for binding to the c-Myc gene promoter, drastically suppresses also the c-Myc gene, reshaping host gene expression profiles to facilitate viral lytic replication (Lee and others 2014). Moreover, since c-IRF4, by competing with c-IRF5, inhibits proinflammatory cytokine production downstream TLR activation (Negishi and others 2005), vIRF4 may also push an inflammatory response.
Thus, as HHV8 employs different encoded proteins in addition to v-IRFs, to contrast innate and adaptive immunity response, the effects of HHV8 vIRFs are not only limited to interference with the IFN response but also interfere with other cellular pathways that impact cell transformation, apoptosis, and autophagy (Lee and others 2012).
The key role of IRF1 in HPV infection is exemplified by the mechanisms used by the virus to both inhibit and exploit it. HPV-16 encodes 2 proteins (E7 and E5) that target IRF1, but in an opposite way. E7 binds to and inactivates IRF1 transcriptional activity by recruiting HDAC to target promoters (Park and others 2000). This results in inhibition of IRF1-mediated induction of both antiviral and antioncogenic genes. The E5 protein that plays an important role in the early phases of tumorigenesis instead does not repress, but rather stimulates IRF1 expression and as a consequence IFN-β and a set of ISGs (Muto and others 2011). Curiously, however, this IFN signature, instead of protecting cells from infection, has been associated with loss of HPV-16 episomes and cervical tumor progression (Pett and others 2006). Thus, depending on the cell type considered and the timing in the viral life cycle, HPV targets IRF1 through different viral proteins and in an opposite way. This results in both favoring immune escape and tumor progression.
In HCV infection, targeting of both IRF1 and IRF7 may similarly favor both viral escape from host immunity and establishment of a chronic infection. As mentioned above, we have reported that IRF1 is inhibited by the core protein (Ciccaglione and others 2007). This inhibition not only silences antiviral but also immunomodulatory genes as IL-12, IL-15, and genes important for antigen presentation such as the immunoproteasome subunit LMP2. IL-12 is known to play a pivotal role in the generation of Th1 immune response and, as indicated by in vivo studies, a Th1 phenotype can clear the virus, while a Th2 dominance tends to favor chronic infection. Core-mediated inhibition of IRF1 may thus be instrumental in downmodulating Th1 responses in favor of Th2 responses.
Interestingly, the HCV core protein inhibits IRF1 promoter activity even after treatment with inflammatory cytokines. This raises the possibility that both locally, in hepatoma cells, and systemically by affecting PBMCs, the core protein, through the repression of IRF1, can also depress the inflammatory response induced by virus infection. Thus, IRF1 repression by the HCV core protein may, at least in part, recapitulate the reported HCV core ability in evading the host response at several levels: antiviral, inflammatory, and immune (Ciccaglione and others 2007).
An impairment in the IFN-alpha signaling pathway due to inhibition of IRF7 nuclear translocation has been similarly suggested as, in part, responsible for establishment of chronic HCV infection (Raychoudhuri and others 2010).
In HIV-1 infection, instead, both IRF1 and IRF7 are exploited by the virus to increase infectivity and pathogenesis. The manipulation of IRF1 in HIV-1 infection represents a paradigm of the HIV-1 ability to have evolved mechanisms to evade host defenses and exploit a host factor to its own advantage. In myeloid cells, by inducing IRF1 expression, HIV-1 stimulates a distinct subset of ISGs that despite displaying intrinsic and unique antiviral actions, does not restrict viral infection (Harman and others 2011). Similarly, we showed that in T cells, IRF1 is induced by HIV-1 and exploited to stimulate LTR-directed viral transcription in the early phases of infection, alone or in combination with suboptimal doses of the viral transactivator, Tat (Sgarbanti and others 2002, 2008).
Interestingly, also extracellular secreted Tat may induce IRF1 expression, thus increasing T-cell activation and creating a favorable environment for viral infection/replication of yet uninfected cells. On the other hand, later in infection, when discrete amounts of Tat are produced and IRF1 activity on LTR is dispensable for the virus to replicate, IRF1 is specifically targeted by Tat that sequesters it, quenching its transcriptional activity on target genes (Remoli and others 2006). Tat also disarms IRF1 by favoring IRF1 ubiquitination and degradation through the proteasome, as mentioned above (Remoli AL, unpublished). Finally, even if not yet formally demonstrated, IRF1 and IRF8 can be exploited by HIV-1 in the establishment and maintenance of the latent state (Sgarbanti and others 2002; Munier and others 2005; Battistini and Sgarbanti 2014).
HIV-1 also exploits IRF7, taking advantage of the key role of IRF7 in IFN production by pDCs. In HIV-1 infection, indeed, the exaggerated production of IFN-α by HIV-activated pDCs in vivo mediated by IRF7 substantially contributes to HIV pathogenesis rather than to protection (Remoli and others 2011; Marsili and others 2012; Boasso 2013; Manches and others 2014; Acchioni and others 2015).
IRF7 is similarly heavily hijacked and exploited by EBV to regulate viral replication and EBV-induced latency and transformation (Ning and others 2011; Perrotti and others 2013). Curiously, the Irf7 gene was originally cloned in the context of latent EBV infection as a factor able to bind the Qp promoter in EBV type III latency (Zhang and Pagano 1997). EBV-encoded LMP1, the major EBV oncoprotein and one of the key viral proteins required for the transformation of primary B cells, induces the expression of IRF7 and activates it through the PRR signaling adaptor, TRAF6, as mentioned above. Moreover, IRF7 induces expression of LMP1 so that it forms a regulatory circuit that may potentiate oncogenic effects of both factors (Ning and others 2003). The induction and binding of IRF7 may indeed cause the silencing of Qp promoter in EBV type III latency, contributing to the maintenance of EBV long-term latency (Zhang and Pagano 2001), and cooperate with LMP-1 in mediating transformation through regulation of a set of genes involved in cell growth and proliferation in EBV-transformed cells. On the other hand, deactivation of IRF7 by LMP1 upon induction of the ubiquitin-editing enzyme A20 (Laherty and others 1992; Ning and Pagano 2010), as mentioned above, highlights the importance of the LMP1/IRF7 interplay in the equilibrium between lytic replication and maintenance of latency.
Other oncogenic IRFs, as IRF2 and IRF4, similarly have been implicated in EBV-induced transformation and are induced and activated by LMP1 (Huye and others 2007; Xu and others 2008). Recently, it has been reported that also the EBV nuclear antigen 3C (EBNA 3C), an encoded latent antigen required for growth transformation of primary human B-lymphocytes, can contribute to B-cell transformation by modulating the molecular interplay between cellular IRF4 and IRF8 (Banerjee and others 2013).
IRF5 (as IRF7) is expressed in higher abundance in EBV latency III and negatively regulates IRF7-mediated induction of LMP1 most likely by heterodimerization with IRF7 in EBV latency (Ning and others 2005). These observations highlight the complexity and still not completely understood consequence of IRF modulation in the context of EBV infection (Ning and others 2011).
Conclusions and Future Perspectives
Viral infections represent an ever-present threat for human beings, with new and/or reemerging pathogens arising all the time, as well illustrated by the recent outbreaks of EBOV and Coronaviruses. To effectively resist this continual threat from the environment, vertebrates have developed several effectual defense mechanisms, including innate and adaptive immunity. For against successful pathogens have evolved sophisticated strategies to evade, subvert, and/or exploit these defenses, where blocking the innate response in the first place is required to replicate and survive.
In recent years, our understanding of the nature of mechanisms involved in the activation of an innate response by pathogens has evolved significantly and, in parallel, specific processes by which pathogenic microorganisms subvert these innate immune pathways are becoming progressively appreciated. These viral anti-immune strategies can be defined as reactive, which directly act on host defenses, and proactive, which induce and/or subvert the host responses to their own benefit. Although the various strategies are numerous and different, there are several general mechanisms that are shared between different viruses and it is not uncommon for a virus to target a signaling pathway at multiple steps using multiple viral proteins, as well as to use a single viral antagonist to target different components of the pathway.
As described, viruses have learned to manipulate host immune control mechanisms by evolving genes to target specific pathways or using captured host genes. The highest degree of multifunctionality and conservation of viral proteins is observed in RNA viruses that (due to their relatively limited genome size) must maximize functions of single proteins, at variance with DNA viruses whose larger genomes may incorporate host genes that may serve as antagonists.
Viral countermeasures include modification and/or the hiding of viral PAMPs, thus eluding detection in the first place and the use of viral proteins that share cellular-like domain or cellular-like enzymatic properties that can compete with or destroy the host counterparts.
In this study, we have compiled a wide-ranging list of known strategies, focusing on those specifically targeting members of the IRF family of transcriptional regulators. IRFs represent a sparkling and versatile family of transcription factors that exert critical roles in the maintenance of homeostasis of mammalian systems. Their importance in immunity is reflected by their striking evolutionary conservation (Nehyba and others 2009) and by the fact that they are prior and compulsive targets of any known pathogen. As described, viral proteins may complex and inactivate both IRF activators and/or IRF themselves, may serve as substrates for IRF activators, and may redirect or mimic active host enzymes that affect IRF stability.
Downstream PRRs, IRFs represent the last step of the signaling pathway in IFN induction and their targeting could appear as a redundant mechanism in the event that a pathogen has successfully already blocked initial detection or upstream molecules in the signaling pathway. However, IRFs are also endowed with intrinsic antiviral activity that allows the host cell to use the IRF-mediated antiviral response also when the virus has circumvented IFN production. In addition, most IRFs specifically regulate hematopoietic differentiation and activation of immune cells and control cell cycle and apoptosis. These activities are also brightly exploited by some viruses to regulate their own replication and survival in a latent state while favoring persistence and, in some cases, progression to cell transformation.
Learning from these viral escape mechanisms may have several translation implications and may help in understanding how to fit out the immune system to cheat the pathogen and to shift the balance in favor of the host.
Future applications include the genetic manipulation of viral IRF antagonists in viral genomes for the development of promising candidates for live vaccines. Many of the pathogen proteins responsible for IRF antagonism are also determinants of virulence and pathogenesis. As such, they are highly conserved and may thus constitute attractive targets for the development of therapeutics against various clinically relevant pathogens, reducing the bias of resistance mutations. In turn, IRFs acting at specific nodes and cross-regulating different pathways might turn out to be new drug targets for treating a range of diseases caused by different viruses.
On the other side of the coin, the recovery of a full IRF activity must be fine-tuned. There is indeed a fine balance between a protective host response and an imbalance that can cause disease. Uncontrolled IRF expression, whatever it impacts on the innate response or on the cell growth control and apoptosis, could play a pathogenic role, as already reported for uncontrolled IFN expression as a determinant of disease progression in some bacterial and viral infections (Malireddi and Kanneganti 2013; Li and others 2014; Acchioni and others 2015; Snell and Brooks 2015). Nevertheless, even in these cases, the lesson learnt from viral strategies that exploit IRF functions could aid in harnessing pathogen proteins themselves or modified versions of them as therapeutics. These could work in suppressing overwhelming immune activation also in nonpathogen-induced diseases that arise from an inappropriate response to host components as autoimmune and inflammatory diseases. This would be a bright way of hijacking molecules evolved during pathogen adaptation and associated fitness to shift the balance to the host advantage.
As yet, interrelationships between the role of IRFs in immunity and other regulatory pathways of metabolism are still an unexplored area of research. To date, little is known on the role of IRFs in metabolic homeostasis, and the involvement in the regulation/interference with some metabolic pathways linked to IRF-mediated immune responses is just emerging, as recently discussed (Perrotti and others 2013; Zhao and others 2015). Future studies, by using system biology strategies, aimed at unraveling the IRF involvement in these pathways as well as the elucidation of the full range of target genes triggered by IRFs and functional deregulation of their activation in both physiological and pathological conditions, will define a holistic understanding of IRF activities with important therapeutic implications.
Finally, ongoing metagenome studies aimed at the characterization of the resident virome/virobiota in mammalian physiology and disease, even if still in their infancy, have already demonstrated that the virobiota is far more than a source of health threat and that some viruses may also function much like symbiotic bacteria. The impact of virobiota on host immunity is just emerging, but understanding the role, either beneficial or detrimental, of the virome/virobiota, besides enormously increasing our knowledge on the most abundant and fastest evolving genetic elements on Earth (Virgin 2014; Cadwell 2015), will surely open new avenues for the next generation of scientists to devise new strategies to manipulate/exploit these, at times, friend viruses to the host benefit, as we are starting to do with bacteria.
Footnotes
Acknowledgments
The authors apologize to all colleagues whose works have not been separately cited or discussed here due to space or knowledge limitations. This work was, in part, supported by institutional grants.
Author Disclosure Statement
No competing financial interests exist.