
Abstract
Toll-like receptors (TLRs) are major receptors of the host innate immune system that recognize conserved pathogen-associated molecular patterns (PAMPs) of invading microbes. Activation of TLR signaling culminates in the expression of multiple genes in a coordinate and kinetically defined manner. In this review, we summarize the current studies describing the chromatin landscape of TLR-responsive inflammatory genes and how changes to this chromatin landscape govern cell type-specific and temporal gene expression. We further elaborate classical endotoxin tolerance and epigenetic mechanisms controlling tolerance and interferon priming effects on inflammatory promoters.
Introduction
I
Among the various families of PRRs (eg,Toll-like receptors [TLRs], Nod-like receptors, RIG-I-like receptors [RLRs], c-type lectin receptors [CLRs], and cytosolic DNA receptors), TLRs are one of the largest and best-studied families of PRRs. PRRs recognize PAMPs or DAMPs, and lead to the activation of specific signaling cascades that converge to induce expression of proinflammatory cytokines, chemokines, and antiviral interferons (IFNs). Thus cell type-specific, dynamically controlled, and appropriately pitched gene expression profile is at the heart of every innate immune response. A growing body of evidence suggests that signaling pathways and transcription factors play a pivotal role; however, epigenetic factors/chromatin-based mechanisms have proved to be critical for context-specific gene expression in diverse innate immune cell types (Saccani and Natoli 2002; Foster and others 2007; Hargreaves and others 2009).
The term epigenetics denotes often heritable changes in gene activity or expression that occur without alternation in the underlying DNA sequence; that is, a change in phenotype without a change in genotype. Genomic DNA in eukaryotic cells is packaged by histones to form protein/DNA complexes termed chromatin. The nucleosome is the basic unit of chromatin, which is composed of 147 bp of DNA wrapped around a histone octamer, consisting of two copies each of four core histones (H2A, H2B, H3, and H4). Linker histone H1 interacts with DNA between nucleosomes and helps to compact chromatin into higher-order structures to form chromosomes. Amino-terminal tails of core histones project out of the nucleosome, making them accessible to histone-modifying enzymes that either add or remove covalent chemical modifications (eg, acetylation, methylation, phosphorylation, sumoylation, and ubiquitination), that influence chromatin organization/compaction and function [reviewed by (Kouzarides 2007)].
Simplistically, covalent modifications either disrupt chromatin contacts, affecting its level of compaction, or modulate recruitment of nonhistone regulatory proteins to chromatin, to influence many fundamental biological processes. Basically, there are three main classes of regulatory proteins: “writers” that create these covalent modifications (eg, DNA and histone methyltranferases, histone acetyltransferases, etc.), “erasers” that eliminate modifications (eg, demethylases, histone deacetylases, etc.), and “readers” that recognize modification through specific protein domains (eg, bromodomain-containing proteins), which are the major players that govern dynamic changes in chromatin structure on the basis of developmental or environmental stimuli [reviewed by (Bannister and Kouzarides 2011; Yun and others 2011)]. In the past few years, there have been spectacular advances in the understanding of epigenetic mechanisms, which predominantly include DNA or histone modifications, histone variants, noncoding RNAs, chromatin architecture (nucleosome occupancy and positioning), and nuclear organization that are reviewed elsewhere in detail [reviewed by (Li and others 2007; Maze and others 2014; Carpenter and Fitzgerald 2015)]. The following sections will synopsize the emerging aspects of current epigenetic research within innate immunity and inflammation utilizing the TLRs as a model PRR system.
Chromatin Dynamics at TLR-Inducible Promoters/Innate Immune-Related Genes
It has long been known that ligation of TLRs or IFN receptors induces a broad and significant transcriptional program that results in the rapid upregulation of hundreds of host genes [reviewed by (Kawai and Akira 2011)]. While there has been tremendous progress over the last decade in describing in great detail the molecular signaling events leading from the TLRs to the promoter regions of inflammatory response genes, far less has been revealed about the role epigenetics plays in shaping the transcriptional responses downstream of the TLRs, as well as other innate immune receptor systems. However, relatively recent work by a number of groups has definitively shown that TLR-induced epigenetic changes constitute a critical new landscape in both the positive and negative regulation of TLR-induced genes.
Transient gene induction is regulated by balanced interplay of transcriptional and chromatin machinery. The transcription cycle of RNA polymerase II (RNA Pol II) is typically divided into four steps: initiation, elongation, termination, and reinitiation, which are highly interconnected and regulated at multiple levels by the defined sequential action of regulatory factors.
Upon activating stimuli, transcription factors bind to the promoters of specific genes and initiate sequential modification events, which result in the expression or silencing of the gene. Initially, recruitment of transcription machinery to form the preinitiation complex was considered as major rate-limiting step by which transcription is regulated. However, several reports have established control of (i) promoter proximal pausing of initiated RNA Pol II and (ii) resumption of transcription elongation, as separate and distinct rate-limiting steps in the induction of immediate early genes (IEGs) like heat shock genes and c-myc (Bentley and Groudine 1986; Gilmour and Lis 1986; Rougvie and Lis 1988; Krumm and others 1992).
Emerging evidence from genome-wide analyses have shown that numerous developmental and stimulus-responsive genes in higher eukaryotes have promoter proximal region-bound transcriptionally engaged RNA Pol II (Guenther and others 2007; Muse and others 2007; Zeitlinger and others 2007; Rahl and others 2010). In macrophages, lipopolysaccharide (LPS) signaling through TLR4 induces several hundred genes, which can be divided into two categories, primary response genes (PRGs) and secondary response genes (SRGs), based on their requirement for new protein synthesis. PRGs are generally induced within an hour of stimulation, whereas the induction of SRGs is delayed due to the requirement for new protein synthesis and chromatin remodeling at their promoters (Ramirez-Carrozzi and others 2006).
In one of the most impactful studies in this area to date, Hargreaves and others carried out an elegant study of both the steady-state and inducible changes in the patterns of histone modification and promoter occupancy of many TLR-responsive inflammatory genes in macrophages (Hargreaves and others 2009). Remarkably, they found that the chromatin surrounding the transcriptional start sites (TSS) of many PRGs (eg, IL-6) shows modifications characteristic of open or permissive chromatin, even in the unstimulated (basal) state. This epigenetic code markedly differentiates PRGs from the indirectly induced SRGs. Reflecting the open chromatin state, some PRGs (eg, IRF-1) have a pronounced basal level of expression that is further enhanced by TLR ligation (Hargreaves and others 2009). In a wide ranging ChIP analysis of RNA pol II-associated genes, many PRGs were found to have significantly elevated basal levels of the histone H3 trimethylated at lysine 4 (H3K4me3) modification surrounding the TSS, usually characteristic of actively transcribed promoters.
In contrast, the SRGs did not show elevated basal levels of H3K4me3 and only inducibly acquired this modification following TLR ligation. Similarly, histone H3 acetylation (H3Ac), another modification typical of actively transcribed promoters, is constitutively identified on PRGs, but not on SRGs (Hargreaves and others 2009). The presence of such elevated basal levels of H3K4me3 and H3Ac were simultaneously shown to correlate with strong GC content/CpG elements in the promoters (Ramirez-Carrozzi and others 2009).
In accordance with the H3K4me3 and H3Ac data, high levels of the RNA pol II enzyme were preassociated with PRG promoters in the basal state, despite a lack of active transcription. It was found by Hargreaves and others that the paused RNA pol II on the promoters of PRGs was phosphorylated on Serine 5 (S5), but not Serine 2 (S2) of the C-terminal domain (CTD), with both phosphorylation events being required to initiate productive transcription elongation. RNA pol II did not acquire S2 modification until after TLR ligation.
As a possible explanation for the basal epigenetic priming of PRGs, it was found that continued association with the SP1 transcription factor at upstream GC-rich/CpG elements, which may be a critical component of the process for marking PRGs for subsequent basal chromatin modification, as knockdown of SP1 expression dramatically reduces steady-state RNA pol II S5 phosphorylation at PRG-I promoters. As an additional consequence of RNA pol II preassociation, PRGs constitutively produce short but unprocessed transcripts that are required for the maintenance of positive histone modifications at these promoters in a positive feedback loop (Hargreaves and others 2009).
Following TLR ligation, the inducible addition of histone H4 acetylation at lysine 5, 8, and 12 (H4K5, H4K8, and H4K12 Ac) at PRG promoters allows binding of the double bromodomain-containing protein BRD4. The epigenetic reader, BRD4, conserved from yeast to humans, recognizes inducible acetylation marks on histones (such as H4K5/8/12 Ac) and certain nonhistone proteins (acetylated RelA) and serves as a scaffold for assembly of multiprotein complexes that regulate transcription (Wu and Chiang 2007; Huang and others 2009). Bound BRD4 facilitates recruitment of positive transcription elongation factor, P-TEFb complex to phosphorylate S2 at CTD of RNA Pol II and thereby permit active transcription. A schematic detailing of steady-state and TLR-induced epigenetic transcriptional mechanisms on early TLR-responsive promoters is shown in Fig. 1A and B.
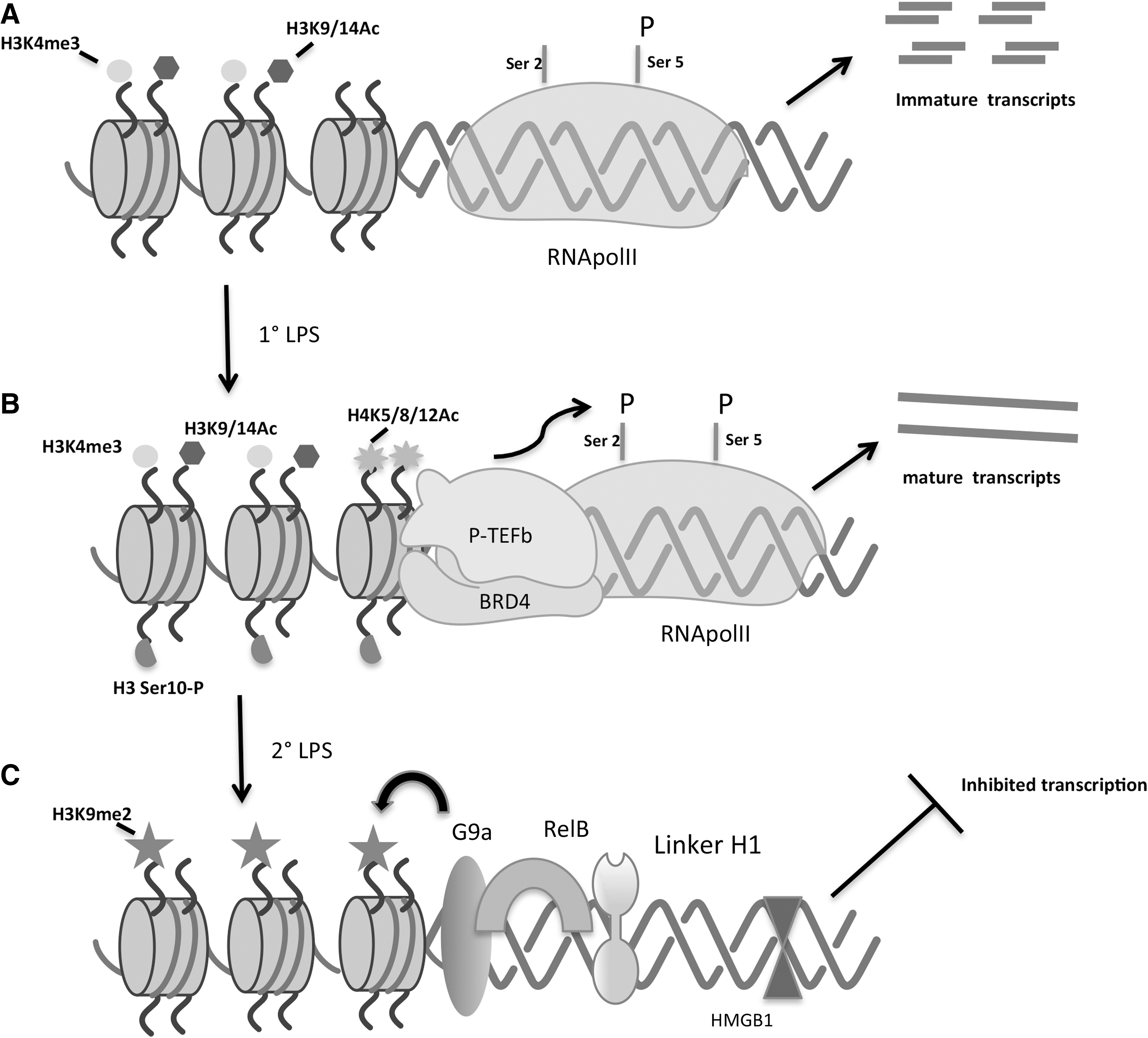
Steady-state and TLR-induced epigenetic transcriptional regulation of an early TLR-responsive promoter
The work of Hargreaves and others was independently recapitulated in work by the Glass group (Escoubet-Lozach and others 2011). This study performed a comprehensive genome-wide analysis of the epigenetic and transcriptional status of TLR4-induced genes and found similar patterns of basal chromatin modification. Specifically, ChIP sequencing of 130 immediate/early (I/E) and 120 late TLR4-regulated genes found that 89% of I/E promoters both in thioglycollate-elicited peritoneal macrophages and bone marrow-derived macrophages (BMDM) showed an elevated H3K4me3 signature. Histone H3 acetylation on lysine 9 and 14 (H3K9/14 Ac) was also found to be present with substantial enrichment on TLR4-inducible I/E promoters (Escoubet-Lozach and others 2011).
A comparison of the H3K4me3 signatures at I/E promoters between macrophages and murine embryonic fibroblasts (MEFs) revealed that macrophage promoters are highly poised to respond to TLR ligands, whereas the same promoters in MEFs are not. Thus, cell type-specific, differential chromatin priming may underlie much of the tissue-specific differences in the inducible TLR gene repertoire (Escoubet-Lozach and others 2011).
Type I IFN induces a distinct subset of several hundred IFN-stimulated genes (ISGs), many of which are crucial to establish a cellular antiviral state (Schoggins and others 2011). Upon IFN stimulation, negative elongation factor (NELF) and 5, 6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) sensitivity-inducing factor (DSIF), which promote the pausing of RNA Pol II, are also recruited to ISGs to restrain transcriptional output, and BRD4 coordinates both positive (mediated by P-TEFb) and negative (mediated by NELF/DSIF) regulation of ISG elongation (Patel and others 2013). S2 phosphorylation of CTD serves as a platform for binding of partner proteins, including elongation factors, RNA processing and termination factors, and chromatin modifiers that travel with RNA Pol II into the gene body to aid in productive elongation (Phatnani and Greenleaf 2006).
In yeast, Set2 methyltransferase is targeted to coding regions by its interaction with S2 phosphorylated RNA Pol II CTD to methylate H3K36 (Krogan and others 2003; Li and others 2003). Di- and trimethylation of histone H3 at lysine 36 (H3K36me2 and H3K36me3) are associated with transcribed genes and considered as an activating histone mark [reviewed by (Kouzarides 2007)]. Interestingly, it is being shown that H3K36me2 and H3K36me3 employ Rpd3S histone deacetylase complex at the coding regions to reset the chromatin behind the elongating polymerase (Keogh and others 2005; Govind and others 2010). However, it is not known whether and how H3K36 methylation is connected to create/add more activating marks on chromatin. In higher eukaryotes, one such activating mark is histone variant H3.3, which is deposited mainly over transcribed regions in a replication-independent manner and has been implicated in the creation of an epigenetic mark to preserve transcriptional memory (Ng and Gurdon 2008).
The histone chaperon HIRA plays an important role in H3.3 deposition over transcriptionally active genes and it has been shown to bind both the initiating and elongating forms of RNA Pol II (Ray-Gallet and others 2011). Recently, it has been identified that histone methyltransferase, Wolf-Hirschhorn syndrome candidate 1 (WHSC1), interacts with BRD4 and P-TEFb to facilitate transcription elongation. On the other hand, WHSC1 also associates with histone variant H3.3-specific chaperone HIRA and is coresident on ISG coding regions and deposits H3.3, leaving a lasting transcriptional mark. Thus, these findings revealed novel, unrecognized mechanisms by which BRD4 driven recruitment of WHSC1 makes a link between transcriptional elongation and H3.3 deposition in ISGs (Sarai and others 2013).
Importance of Chromatin Regulation in Circumstances of Microbial “Tolerance”
While significant work has been done to characterize the steady-state chromatin landscape that characterizes TLR-responsive genes, as much if not greater work has been done to identify and detail the role that chromatin remodeling and chromatin remodeling elements play in the negative regulation of inflammation that is key to prevent inflammation-related pathologies.
It has long been recognized that monocytes and macrophages from septic patients display a profound loss of responsiveness to ex vivo stimulation by microbial constituents. This state of refractoriness could be recapitulated in culture through exposure of monocyte/macrophages to purified bacterial endotoxin, LPS outer membrane component of Gram-negative bacteria (Cavaillon and Adib-Conquy 2005; Cavaillon and others 2005). One of the molecular hallmarks of LPS-tolerized cells is a greatly diminished transcriptional induction seen at the promoters of LPS-inducible, classical inflammatory genes (Cavaillon and others 2005). Using the IL-1β promoter and THP-1 cells as a model, Chan and others were among the first to associate the loss of LPS-inducible IL-1β transcription in LPS-tolerized THP-1 cells with a differential chromatin architecture (Chan and others 2005).
Initial observations pointing in this direction included experiments demonstrating that while NF-κB p65 transcription factor accumulated in the nucleus to a similar degree, p65 failed to bind to the IL-1β promoter in tolerized monocytes, (Chan and others 2005), suggesting a loss of chromatin accessibility. Subsequent characterization revealed differential histone tail posttranslational modification at the IL-1β promoter in cells rendered tolerant. Such changes included a profound loss of LPS-induced histone 3 serine 10 phosphorylation (H3(S10)P), an event known to positively regulate gene transcription. Similarly noted was a loss of LPS-induced H3K9 and H3K14Ac modifications (Chan and others 2005). In a series of studies investigating the underlying basis for the differential histone modifications, McCall and colleagues discovered a highly ordered series of events resulting in the assembly of repressor complexes at IL-1β and TNF-α promoters in endotoxin-tolerized (ET) THP-1 cells. They found that the level of NF-κB family member RelB is highly elevated in ET cells, RelB remains associated with tolerized inflammatory gene promoters, and that knockdown of RelB can partially restore promoter inducibility (Yoza and others 2006).
It was subsequently reported that the recruitment of RelB to the IL-1β and TNFα promoters depends upon and is preceded by recruitment of the linker histone H1 and the DNA-binding protein HMGB1. Interestingly, knockdown of either linker H1 or HMGB1 could prevent recruitment of RelB and significantly restore promoter inducibility in ET cells (El Gazzar and others 2009). This group has also identified a recruited enzymatic activity of the histone H3 lysine methyltransferase G9a that is physically associated with the RHD domain of RelB and is known to catalyze repressive H3K9 methylation (El Gazzar and others 2008, 2010). Characteristics associated with ET promoters are schematically outlined in Fig. 1C.
A later genome-wide analysis of promoter architecture regulation in ET identified many of the previously reported patterns while adding new insight (Foster and others 2007). A global transcriptomic analysis by Foster and others revealed a trend toward the preferential tolerization of proinflammatory genes over antimicrobial genes. However, an additional investigation into the molecular mechanism of discrimination between tolerized and nontolerized (T vs. NT) genes did not reveal a role for differential signal transduction or promoter transcription factor-binding elements in the T versus NT discrimination (Foster and others 2007). Additionally, roles for the glucocorticoid receptor as well as regulated mRNA stability were ruled out. Using the genes for IL-6 and the formyl peptide receptor (FPR1) as models of tolerizable and nontolerizable genes, respectively, Foster and others developed the hypothesis that gene-specific regulation and discrimination of the class T and NT genes was achieved through promoter-specific chromatin remodeling. They showed directly that RNA pol II was differentially recruited to the IL-6 and FPR1 promoters in tolerized macrophages (Foster and others 2007). Specifically, they found that the H4Ac modification found inducibly on TLR4-driven genes during primary LPS stimulation, was lost on all TLR4-induced genes following LPS stimulation, and only reacquired at the NT promoters. In fact, the H4Ac status was reacquired on NT promoters at a higher resting state than observed before the initial LPS priming. It was further identified that Trichostatin A, a deacetylase inhibitor, could substantially reverse the suppression of IL-6 and restore H4Ac in tolerized cells.
Foster and others also showed that H3K4me3 inducibility occurs at promoters following TLR4 ligation and is selectively lost at tolerized promoters. Pargyline, an LSD1 demethylase inhibitor, could prevent IL-6 silencing in tolerant macrophages.
Mechanistically, the positively acting chromatin remodeling complexes BRG-1 and mi-2β are inducibly recruited to TLR-responsive promoters and this recruitment is lost in tolerance at ET promoters.
Interestingly, experiments using the DRB elongase inhibitor/RNA Pol II inhibitor revealed that TLR-induced genes were required for the subsequent negative chromatin remodeling at ET promoters, but the nature of these TLR-induced elements remains unknown to this day.
One interesting point of biology to emerge from the Foster and others' study is that they did not detect H3K9 or H3K27 methylation on tolerant gene promoters, while finding numerous other negative modifications, indicating the selective use of negatively acting histone modification in tolerance.
The relevance of G9a as a specific mediator of chromatin regulation in tolerance was confirmed by a systems biology approach by Chen and colleagues (Liu and others 2014). Utilizing a novel chemoproteomic protocol involving a small-molecule competitive inhibitor of the enzymatically active form of the G9a, H3K9me2 methyl transferase, this group identified a large multicomponent “repressome” complex regulating chromatin at the promoters of LPS-tolerized genes. Compellingly, it was shown that G9a not only coordinates deposition of combinatorial methyl-lysine codes on histone H3, but also stabilizes select transcription factors to promote their corepressor activity. Specifically, G9a is required to recruit and stabilize c-Myc at promoters that drive the gene-specific repressive functions of Myc in tolerance (Liu and others 2014).
Utilizing a similar systems interactome methodology, the Chen group identified the catalytic unit of phosphatase 2A (PP2Ac) as a tolerance-specific MyD88-interacting partner (Xie and others 2013). Knockdown of PP2Ac partially rescued expression of some tolerized genes (eg, IL6, CCL2, and CCL17) in RAW 264.7 murine macrophage-like cells. Unexpectedly, association with PP2A was found to facilitate MyD88 translocation to the nucleus following TLR ligation. Phosphoproteome profiling revealed components of the SWI/SNF remodeling complex as nuclear targets for PP2A. Knockdown of PP2Ac prevented a loss of H3Ser10 phosphorylation in tolerance that was dependent upon PP2Ac association with MyD88. Similarly, PP2A was found to strongly influence MyD88-dependent histone deacetylation and nucleosome remodeling in tolerance (Xie and others 2013).
Innate receptor-induced negative chromatin regulation occurs not only in instances of TLR homotolerance, but has also been shown to be critical in mediating inflammation in instances of TNFR-TLR cross-tolerance. In this instance, TNF-α-induced remodeling of the IL-6 promoter was shown to be dependent on the activity of GSK3 kinase (Park and others 2011).
The Interferons as Educators of the Chromatin Landscape at Inflammatory Promoters
Each of the IFN systems described in mammals has a potent capacity to directly induce hundreds of genes, many of which have been described to be directly antimicrobial (Schoggins and others 2011). Additionally, the IFNs can have profound effects on the transcriptional programs induced by ligation of secondary innate immune receptors. For many years it was believed that these priming actions of the IFNs were achieved by modulating expression and/or posttranslational modification of relevant transcription factors. However, one of the most exciting areas of IFN research presently entails a description of the effects that autocrine and paracrine exposure to IFNs may have on the chromatin modification pattern at important pro- and anti-inflammatory promoters.
Type I interferons
Type I IFNs are primarily associated with potent innate-driven antiviral defense. In this capacity, type I IFNs transcriptionally upregulate hundreds of genes through the STAT family of transcription factors (Schoggins and others 2011). A role for type I IFN in mediating chromatin remodeling has also recently been described (Schliehe and others 2015). It was shown that the local production of type I IFN upregulates the transcription of a single lysine methyltransferase SETDB2. SETDB2 is a member of the SUV39 family of methyltransferases and is known to catalyze the trimethylation of H3K9 at select promoters (Xu and others 2010). The generation of a hypomorphic mouse line revealed that type I IFN-induced SETDB2 catalyzes the addition of H3K9me3 modifications on the promoters of a subset of NF-κB responsive genes, including the neutrophil chemokine CXCL1.
In a mouse model of influenza and Gram-positive bacterial coinfection, it was shown that SETDB2-mediated repression of inflammation led to increased bacterial replication and pathology in the lungs of superinfected animals (Schliehe and others 2015). Whether SETDB2 is the only type I IFN-induced chromatin-modifying enzyme remains to be determined, as well as the full extent of SETDB2 target genes.
Type II interferon—IFN-γ
Type II IFN (IFN-γ) has been shown in many studies to increase the antimicrobial functions of macrophages (Schroder and others 2004). The treatment or priming of macrophages with IFN-γ does not directly induce cytokines, but dramatically enhances the subsequent production of TNF-α, IL-6, and IL-12p40 mRNA during bacterial-induced TLR ligation (Hu and Ivashkiv 2009).
Qiao and others found that IFN-γ priming of murine macrophages significantly increases RNA pol II recruitment to TNF-α, IL-6, and IL-12 p40 promoters (Qiao and others 2013). Additionally IFN-γ prolongs the binding of NF-κB constituents to inflammatory promoters. Nucleosome remodeling (as judged by restriction enzyme accessibility assay) is also enhanced as a result of IFN-γ. Basal as well as induced levels of H4Ac are increased (Qiao and others 2013).
Strikingly, ChIP Seq revealed that IFN-γ priming of murine macrophages induces a dramatic change in the density of H3K27Ac tag at individual gene loci. IFN-γ priming had a much more pervasive effect on genomic distribution of H3K27Ac peaks than did the LPS stimulation. Specifically, IFN-γ induced broad H3K27Ac peaks 25 kb, 50 kb, and 65 kb upstream of the IL-6 TSS, an area that overlaps with previously identified sites of increased DNAse I hypersensitivity (Qiao and others 2013).
This same study found unexpectedly that STAT1 was recruited to the promoters of TNF-α, IL-6, and IL-12 p40 promoters following IFN-γ treatment, despite the absence of canonical STAT1 sites. Correspondingly, the peak H3K27Ac occurs near STAT1-binding sites suggesting a causal relationship (Qiao and others 2013).
An important conclusion of this work is that cytokine-activated STAT1 might not function solely as a signaling transcription factor, but may also act to initiate chromatin remodeling to alter epigenetic traits.
Finally, another relevant biological role for type II IFN in epigenetics is its ability to substantially reverse the chromatin remodeling found in endotoxin tolerance (Docke and others 1997; Randow and others 1997; Chen and Ivashkiv 2010).
JMJD3 and Chromatin Regulation in Macrophage Functional Polarization
The functional polarization of human and murine macrophages into the classical or alternatively activated states (M1 or M2 activation, respectively) is critical to orchestrate the appropriate innate and adaptive immune response to different classes of pathogens (Murray and others 2014). The expression of the jumanji-C (jmjc) domain containing protein JMJD3 has been known to be upregulated in a TLR-dependent fashion through NF-κB (De Santa and others 2007). Satoh and others have described a role for JMJD3 in regulating M2 macrophage polarization through chromatin remodeling (Satoh and others 2010).
Generation of JMJD3 knockout mice revealed that JMJD3 was dispensable for in vitro inflammatory responses to all known TLR ligands. However, the expression of classical M2 markers on macrophages elicited by intraperitoneal injection of chitin in the JMJD3−/− mice was severely impaired. It was subsequently revealed that immunity to the parasite Nippostrongylus brasiliensis was significantly impaired as was N. brasiliensis recruitment of M2 polarized macrophages into the lung. JMJD3 was previously reported to posses H3K27 methylase activity and a genome-wide screen for promoter effects of JMJD3 expression on H3K27 trimethylation revealed IRF4 as a potential target of JMJD3 activity. IRF4 induction in response to parasite infection was severely inhibited in the JMJD3 knockout animals and ectopic expression of IRF4 was sufficient to restore M2 marker expression in the absence of JMJD3 (Satoh and others 2010).
Noncoding RNA
Another area of recent rapid development is the discovery of long noncoding RNA (lncRNA) and its role as important regulators of gene expression of innate immunity [reviewed in (Carpenter and Fitzgerald 2015)]. LncRNA is larger than 200 nucleotides in length and does not encode proteins. Some of the earlier studies noted that lncRNAs were differentially regulated in virus-infected cells and in DC following LPS stimulation (Guttman and others 2009; Peng and others 2010).
In 2013, Carpenter and others reported that lncRNA-Cox2, an lncRNA product of the gene located adjacent to the prostaglandin-endoperoxide synthase 2 (Ptgs2/Cox2) gene, is highly inducible in macrophages and DCs upon TLR ligand treatment and further showed that lncRNA-Cox2 is essential to maintain basal levels of ISGs through interactions with hnRNA-A/B and A2/B1. LncRNA-Cox2 also controls inducible expression of proinflammatory cytokines following microbial challenge, however the exact mechanisms are not yet known (Carpenter and others 2013). Some of the recent discovery of long noncoding RNA regulating innate immune gene expression is described elsewhere in detail [reviewed in (Carpenter and Fitzgerald 2015)].
Concluding Remarks
The pace of discovery within the area of epigenetics and innate immune inflammation continues to quicken and there will doubtless be many critical discoveries within the next couple of years. Among the significant questions to be answered are the molecular means by which general chromatin writer enzymes are targeted to the promoters of individual inflammatory genes with exquisite specificity, as seen in cases of endotoxin tolerance.
There are established evidences to show that type I and II IFNs modulate chromatin architecture at inflammatory promoters. While the type III IFNs bear many similarities in biological function to type I IFNs (Wack and others 2015), presently nothing is known of their effect at inflammatory promoters, which may be one of the exciting area to be explored in future.
The discovery and optimization of small-molecule inhibitors targeting chromatin machinery is another major focus of current epigenetic field. Small-molecule inhibitors of chromatin modifiers, such as histone deacetylases, DNA methyltransferases, and histone methyl transferases, are used in cancer treatment and bear promise for broader and more diverse medical applications [reviewed in (Finley and Copeland 2014)]. Many recent studies provide a proof of principle that BET bromodomain inhibitors have promising therapeutic utility to suppress inflammation (Nicodeme and others 2010; Chan and others 2015). Preliminary scientific assessment of molecular mechanisms targeted by these drugs and attempts to predict their impact on the course of disease in animal models is of critical importance for subsequent clinical trials. It is noteworthy, that JQ1 the derivative of GSK525762 has entered clinical trials, to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity in NUT midline carcinoma and other cancers (Clinical trial # NCT01587703).
Footnotes
Acknowledgments
This work was supported by NIH AI18797 (S.V.) and AI104541 (S.V. and J.B.).
Author Disclosure Statement
No competing financial interests exist.