
Abstract
Because of its tumor-suppressive effect, interferon-based therapy has been used for the treatment of melanoma. However, limited data are available regarding the antitumor effects of pegylated interferons, either alone or in combination with approved anticancer drugs. We report that treatment of human WM-266-4 melanoma cells with peginterferon beta-1a induced apoptotic markers. Additionally, peginterferon beta-1a significantly inhibited the growth of human SK-MEL-1, A-375, and WM-266-4 melanoma xenografts established in immunocompromised mice. Peginterferon beta-1a regressed large, established WM-266-4 xenografts in nude mice. Treatment of SK-MEL-1 tumor-bearing mice with a combination of peginterferon beta-1a and the MEK inhibitor PD325901 ((R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodophenylamino)benzamide) significantly improved tumor growth inhibition compared with either agent alone. Examination of the antitumor activity of peginterferon beta-1a in combination with approved anticancer drugs in breast and renal carcinomas revealed improved antitumor activity in these preclinical xenograft models, as did the combination of peginterferon beta-1a and bevacizumab in a colon carcinoma xenograft model.
Introduction
I
Similar to other small protein drugs, unmodified type I interferons have relatively short circulating half-lives that may require frequent dosing and that may result in limited efficacy. Pegylation, the process of modifying molecules by attaching poly (ethylene glycol) (PEG), has been used to improve the pharmacokinetics (PK) and efficacy of interferons such as pegylated interferon alpha-2a and interferon alpha-2b (Kozlowski and others 2001; Harris and Chess 2003). Pegylation of interferon alpha-2b with a linear 12 kDa PEG increased its half-life (t1/2) from 4–7 h to 27–39 h, and the elimination t1/2 of interferon alpha-2a pegylated with a branched 40 kDa PEG was increased from 3–8 h to 65 h (Glue and others 2000; Rajender and others 2002). In both cases, enhanced antiviral responses against hepatitis C virus and a concomitant reduction of dosing frequency from three times per week to once per week was achieved.
Similarly, pegylation of interferon beta-1a with a linear 20 kDa PEG increased its t1/2, allowing for a reduction in dosing frequency from once per week to once every other week without impacting its efficacy in relapsing remitting multiple sclerosis (Kieseier and Calabresi 2012; Calabresi and others 2014). In addition, recombinant granulocyte colony-stimulating factor (G-CSF) pegylated with a linear 20 kDa PEG had an increased t1/2, resulting in a significant reduction in dosing frequency compared with unmodified G-CSF for the treatment of chemotherapy-induced neutropenia (Crawford 2002). Moreover, pegylation may reduce the immunogenicity of protein therapeutics (Basu and others 2006; Hu and others 2012; Kieseier and Calabresi 2012).
Due to its antitumor activities, interferon-based therapy is used to treat melanoma, renal cell carcinoma, and myeloproliferative disorders (Bracarda and others 2010). Treatment with type I interferons has also shown promising results for metastatic breast cancer (Recchia and others 2009), but not in colon cancer (Schippinger and others 2005). For melanoma, the National Comprehensive Cancer Network guidelines state that pegylated interferon alpha-2b is an appropriate option for patients with completely resected stage III disease, with either positive sentinel nodes or clinically positive nodes (National Comprehensive Cancer Network 2015). However, despite the extensive use of interferons in the treatment of several cancers, additional studies are needed to determine optimal dose and duration of treatment, and novel strategies are needed to decrease toxicity.
Limited preclinical data are available regarding the antitumorigenic effects of pegylated interferon beta. In one study, peginterferon beta-1a was found to reduce the number of radially oriented blood vessels entering SK-MEL-1 melanoma xenografts in nude mice (Baker and others 2006), whereas in another study, pegylated interferon beta-1b resulted in complete regression of NIH:OVCAR3 ovarian tumors grown in nude mice compared with the unmodified protein (Betaseron®), which induced tumor growth inhibition (Lee and others 2013). These results suggest that a pegylated form of interferon beta may be a promising therapeutic option for certain cancers. In particular, combination treatment of peginterferon beta-1a with small molecule anticancer drugs may provide additive or synergistic antitumor effects and/or overcome resistance to these inhibitors. For example, combination therapy of the B-Raf kinase inhibitor dabrafenib and the MEK kinase inhibitor trametinib in patients with BRAF V600 mutation-positive metastatic melanomas circumvented resistance to the B-Raf kinase inhibitor (Flaherty and others 2012).
Given that peginterferon beta-1a has been shown to inhibit tumor blood vessel formation (Baker and others 2006), it is possible that combining B-Raf or MEK kinase inhibitors with peginterferon beta-1a may provide additive or synergistic antitumor activity in melanoma tumors. Similarly, combination of peginterferon beta-1a with paclitaxel (Taxol® prescribing information 2011), bevacizumab (Avastin® prescribing information 2014), Sorafenib (Nexavar® prescribing information 2010), or irinotecan (Irinotecan® prescribing information 2014) may improve the antitumor activity of these approved therapeutics in other cancers. To test this hypothesis, we investigated the effectiveness of peginterferon beta-1a alone and in combination with the MEK kinase inhibitor PD325901 ((R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodophenylamino)benzamide), and other, approved, antitumor drugs in various murine xenograft models, including human melanoma, and human breast, renal, and colon carcinomas.
Due to the species specificity of human interferon beta-1a for human cells (Qin and others 2001; Arduini and others 2004), any antitumor activity observed for peginterferon beta-1a was expected to be mediated as a direct effect upon the human tumor xenografts or potentially an indirect inhibitory effect on tumor-stimulated angiogenesis, for example, inhibition of tumor produced vascular endothelial growth factor (VEGF). Moreover, any potential role of immune effects on the human tumors would not be expected to be manifested in these mouse models because of this species specificity coupled with the fact that the nude and severe combined immunodeficiency (SCID) mice used were immune compromised. The importance of these factors in the interpretation of the results and the potential use of peginterferon beta-1a for the treatment of human cancers is described in more detail in the Discussion section.
Materials and Methods
Cell lines, animals, and reagents
The human SK-MEL-1 melanoma (constitutively active B-Raf V600E mutant), WM-266-4 melanoma (constitutively active B-Raf V600D mutant), A-375 melanoma (constitutively active B-Raf V600E mutant), MDA-MB-468 breast carcinoma, SK-MEL-2 (wild-type B-Raf; Ras Q61R), SK-MEL-5 (constitutively active B-Raf V600E mutant), and MeWo (wild-type B-Raf) were obtained from ATCC and maintained in the recommended culture media and conditions. The human SN12-C renal carcinoma, MDA-MB-231 breast carcinoma, and SW-620 colon carcinoma cell lines were obtained from the National Cancer Institute and likewise maintained. Male athymic nude homozygous (nu/nu) NCr mice were obtained from the Athymic Animal and Xenograft Core Facility (Case Western Reserve University, Cleveland, OH, USA). Female homozygous (nu/nu) mice (Crl:Nu-Foxn1nu ) and CB17 SCID mice (C.BKa-Ighb /IcrCrl) were obtained from Charles River Laboratories (Wilmington, MA, USA and San Diego, CA, USA).
The MEK kinase inhibitor PD325901 was obtained from Sunesis Pharmaceuticals (San Francisco, CA, USA), its vehicle control (PEG400) was from Fisher Scientific (Pittsburgh, PA, USA), sorafenib was from Onyx Scientific (NJ, USA), bevacizumab was from Roche (Nutley, NJ, USA), temsirolimus was from Wyeth Pharmaceuticals (Pfizer, New York, NY, USA), paclitaxel was from Bedford Laboratories (Bedford, OH, USA), and irinotecan was from Sandoz (Holzkirchen, Germany).
Antitumor activity studies in tumor-bearing mice
All in vivo procedures were performed in accordance with Biogen or Cleveland Clinic Institutional Animal Care and Use Committee guidelines. Athymic nude homozygous mice were inoculated subcutaneously (SC) in the flanks with 2 × 106 SK-MEL-1, WM-266-4, A-375, or MDA-MB-468 cells, and when the tumors reached a predetermined size, the tumor-bearing mice were treated for 3 to 7 weeks with various agents, either alone or in combination. Female CB17 SCID mice were inoculated with 5 × 106 MDA-MB-231 cells, 2 × 106 SN12-C cells, or 1 × 106 SW-620 cells, tumors were allowed to grow to a predetermined size, and the tumor-bearing mice treated for up to 7 weeks with various agents, as above. Tumors were measured using calipers, and tumor volume was calculated in all studies using the formula 0.5 × (L × W2), where L = length and W = width with the exception of the studies measuring SK-MEL-1 tumors, where the formula used was πLW2/6, where W = width (minor diameter) and L = length (major diameter).
For peginterferon beta-1a, the dose is expressed as the mass of the protein per kilogram of mouse weight, where the mass of peginterferon beta-1a is the mass of the protein portion only and does not take into account the mass of the attached PEG group (Baker and others 2006).
Immunoblotting and antibodies
WM-266-4 cells were treated with peginterferon beta-1a for 24 h in complete media. Subsequently, cells were washed with phosphate-buffered saline before lysis in Cell Signaling Lysis Buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL Leupeptin; Cell Signaling Technologies, Danvers, MA, USA) containing protease and phosphatase inhibitors (Sigma, St. Louis, MO, USA) for 30 min on ice. Insoluble material was removed by centrifugation. Protein concentration was quantified using the DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Equal amounts of protein were subjected to SDS-PAGE on 4%–12% polyacrylamide gels and transferred to nitrocellulose membranes (Life Technologies, Carlsbad, CA, USA).
For immunoblotting, antibodies included: anti-STAT1, anti-phospho-STAT1 (Tyr701), anti-STAT3, anti-phospho-STAT3 (Tyr705), antitumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), anti-caspase 3, anti-caspase 8, anti-caspase 9 (Cell Signaling Technologies), and anti-actin (Santa Cruz Biotechnology, Dallas, TX, USA). Secondary horseradish peroxidase-conjugated antibodies were obtained from Cell Signaling Technologies. Chemiluminescence was visualized using Kodak MR Film (Thermo Fisher Scientific, Waltham, MA, USA).
Cell viability assay
Cells (2000 per well) were plated in clear bottom 96-well plates in 100 μL complete media and incubated overnight at 37°C/5% CO2. The next day, cells were treated with peginterferon beta-1a in fresh complete media and incubated for an additional 5 days. Cell viability was assessed using the CellTiter-Blue Reagent (Promega, Madison, WI, USA). Briefly, 20 μL of CellTiter-Blue Reagent was added to each well and incubated for 1 h, and the resulting fluorescence was measured at 560/590 nm (Spectra Max; Molecular Devices, Sunnyvale, CA, USA).
Pharmacokinetic analysis
Non-tumor-bearing nude mice were treated with 0.1 mL of 0.2 mg/mL peginterferon beta-1a (20 μg), and blood samples were collected at 0.25, 0.5, 1, 4, 8, 16, 24, 30, 48, 54, and 72 h postdose (n = 3 per time point). The serum concentration of peginterferon beta-1a was determined using a cytopathic effect bioassay that measures the ability of the protein to protect human lung carcinoma A549 cells challenged with encephalomyocarditis (EMC) virus. For the assay, 1.25 × 104 A549 cells in 75 μL Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 2 mM L-glutamine were added to 96-well microtiter plate wells and incubated for 5 to 6 h at 37°C/5% CO2. Starting with a 5-fold dilution, serum samples were diluted serially and added across the plate. Cells were incubated for 24 h, medium was removed, and EMC virus was added. The cells were incubated for 48 h, and 20 μL of CellTiter 96 AQueous One Solution Cell Proliferation Assay Reagent (Promega) was added per well. After 45 to 90 min of incubation at 37°C/5% CO2, plates were read at 490 nm and data were analyzed using four-parameter curve fitting with SoftMax Pro software (Molecular Devices), where the serum concentration of peginterferon beta-1a was determined using a standard curve constructed using known amounts of protein.
To calculate PK parameters, the mean serum concentration data were analyzed using a noncompartmental analysis extravascular input model (Model 200) with WinNonlin version 5.0.1 software (Pharsight, Inc., Mountain View, CA, USA). For simulation, a one-compartment model with first-order kinetics was used to derive PK parameters.
Mouse weight
Mice were weighed throughout each study to determine whether treatment caused overt toxicity that would manifest as weight loss.
Statistical analyses
All statistical analyses were carried out using SAS software (Cary, NC, USA) version 9.2. Longitudinal mixed effect analysis of covariance with the MIXED procedure was used to evaluate changes in response measures over time and to appropriately adjust the correlation of repeated measurements on individual animals. All tumor volumes were subjected to a logarithmic transformation to accommodate the models’ normality assumption. No transformations were applied to mouse weight data. Repeated measures analysis of variance was used to assess the differences between the effect of peginterferon beta-1a or the MEK kinase inhibitor PD325901 compared with the vehicle control.
Results
Effect of peginterferon beta-1a on apoptosis in WM-266-4 melanoma cells
To assess the inhibition of cell growth by peginterferon beta-1a, the viability of SK-MEL-1, SK-MEL-2, SK-MEL-5, MeWo, and WM-266-4 tumor cells treated with peginterferon beta-1a (0.001–1000 ng/mL) was analyzed using CellTiter-Blue Reagent (Promega). SK-MEL-1 cells were resistant to treatment with peginterferon beta-1a, whereas WM-266-4 cells were the most susceptible to the antiproliferative activity of peginterferon beta-1a with an IC50 of 2 to 3 ng/mL (Fig. 1A). To determine whether apoptosis was likely to contribute to the antiproliferative activity of peginterferon beta-1a in WM-266-4 cells, the presence of apoptosis-related markers (TRAIL, caspases 3, 8, and 9) and the DNA repair enzyme, poly (ADP-ribose) polymerase (PARP), was assessed. In addition, STAT1 and STAT3 were evaluated as canonical response markers of type I interferon activity.

Peginterferon beta-1a inhibits the growth of human WM-266-4 melanoma cells in vitro and induces apoptotic markers.
Treatment with peginterferon beta-1a at 10, 100, and 1,000 ng/mL, concentrations that caused significant inhibition of cellular proliferation, resulted in the cleavage of PARP, caspase-8, and -9, and induction of TRAIL (Fig. 1B). Phosphorylation of STAT1 was induced following treatment with peginterferon beta-1a at all concentrations tested, whereas STAT3 phosphorylation was weakly induced at 100 and 1,000 ng/mL of peginterferon beta-1a (Fig. 1B).
PK analysis of peginterferon beta-1a and dose selection
The PK parameters of peginterferon beta-1a administered SC in non-tumor-bearing nude mice were evaluated to assess the correlation between exposure of peginterferon beta-1a, the dosing frequencies used, and the effectiveness of peginterferon beta-1a. Serum concentrations of peginterferon beta-1a demonstrated a t1/2 of 8.5 h. The area under the curve from time zero to infinity (AUCinf) was 42,300 h × ng/mL, the maximum concentration (Cmax) was 5,600 ng/mL, and the time to maximum concentration (Tmax) was 4 h. Modeling of mean serum concentration data using a one-compartment model with first-order absorption and with no lag time indicated that peginterferon beta-1a dosed at 0.8 mg/kg three times a week (TIW) provided the highest drug exposure, twice weekly (BIW) dosing provided close to complete drug coverage over the dosing period (ie, drug was in circulation during the entire testing period), and once weekly (QW) dosing provided the least drug exposure, with a drug-free period of ∼3 to 4 days between doses. Based on the modeling results and preliminary murine xenograft data, BIW dosing was selected as the primary dose frequency.
Effect of peginterferon beta-1a on the growth of human SK-MEL-1 and A-375 melanoma xenografts in nude mice
The antitumor effect of peginterferon beta-1a was assessed in SK-MEL-1 and A-375 melanoma xenografts in nude mice. Peginterferon beta-1a treatment at 0.4 mg/kg significantly inhibited SK-MEL-1 tumor growth (n = 10) relative to vehicle (P < 0.0001; Fig. 2A). For A-375 melanoma tumors, 1.6 mg/kg peginterferon beta-1a showed significant inhibition of tumor growth (n = 10) compared with the vehicle control (SC BIW for 4 weeks; P < 0.0001); the 0.1, 0.2, and 0.4 mg/kg peginterferon beta-1a groups (P < 0.0001); and to the 0.8 mg/kg peginterferon beta-1a group (P = 0.0003; Fig. 2B). Treatment with 0.8 mg/kg peginterferon beta-1a significantly inhibited tumor growth compared with the vehicle control (P = 0.005). However, there was no significant difference in tumor growth inhibition between the 0.8 mg/kg peginterferon beta-1a group and any of the lower doses, nor was there a significant difference among the 0.1, 0.2, or 0.4 mg/kg peginterferon beta-1a groups or in comparison with the vehicle control group (Fig. 2B).
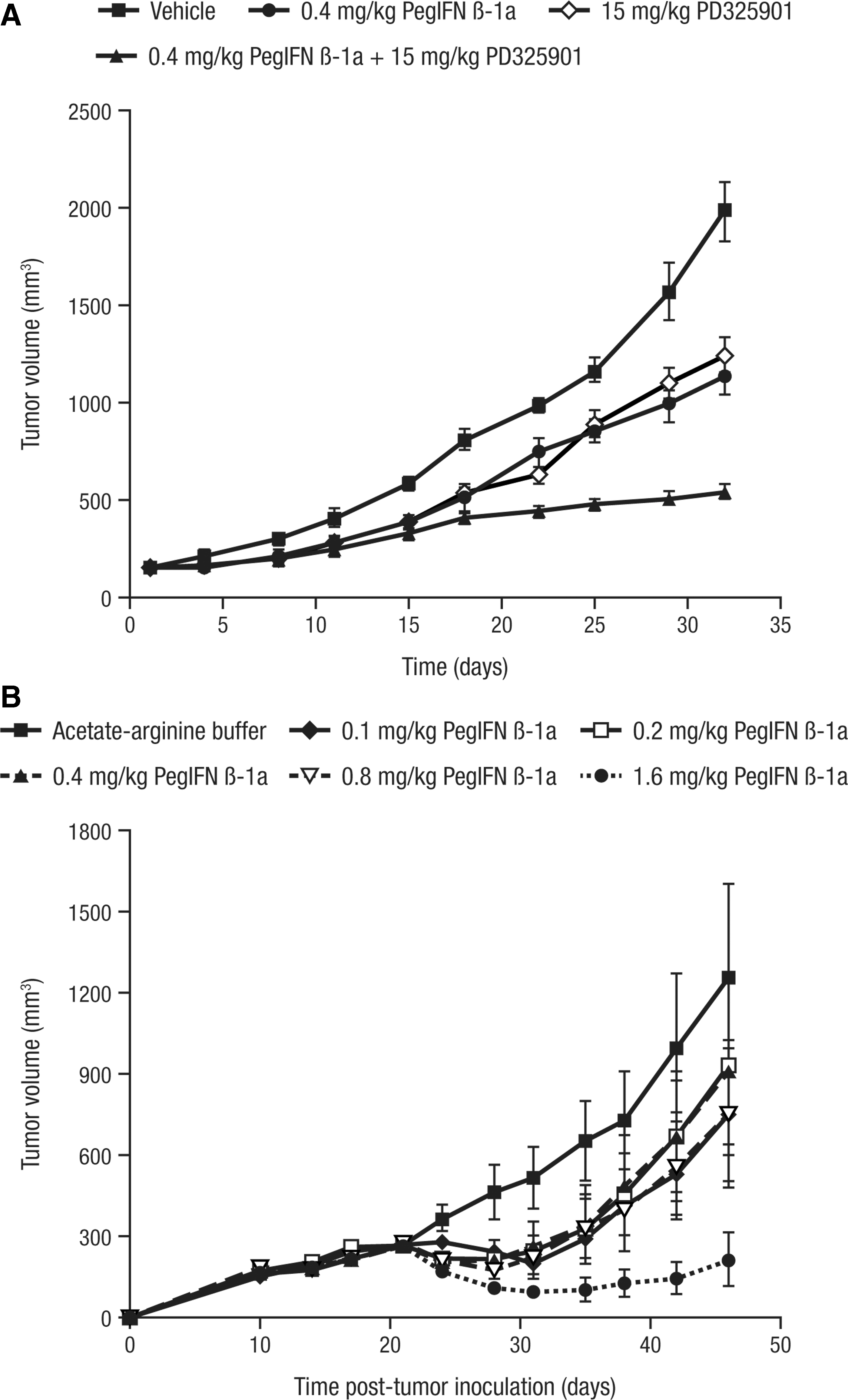
Peginterferon beta-1a inhibits the growth of human SK-MEL-1 and A-375 melanoma xenografts in nude mice.
Effect of peginterferon beta-1a on the growth of human WM-266-4 melanoma xenografts in nude mice
To determine the effect of peginterferon beta-1a dose and dosing frequency on its growth inhibitory activity against WM-266-4 melanoma tumors, nude mice (n = 10) were treated with 0.4, 0.8, or 1.6 mg/kg peginterferon beta-1a QW, BIW, or TIW each for 4 weeks SC. At all dose and dosing frequency combinations, peginterferon beta-1a significantly inhibited melanoma growth relative to the vehicle control (P < 0.0123; Fig. 3). In particular, the QW dose of 1.6 mg/kg peginterferon beta-1a and all doses given BIW and TIW induced tumor regression, with the 1.6 mg/kg QW dose inducing significant tumor inhibition relative to 0.8 mg/kg QW (P < 0.0001). However, no differences were observed in tumor inhibition induced by 0.4 or 0.8 mg/kg peginterferon beta-1a given QW (P = 0.9; Fig. 3A). In mice treated with peginterferon beta-1a BIW and TIW, all three doses tested induced significant tumor regression compared with the vehicle control (P < 0.0001; Fig. 3B, C).
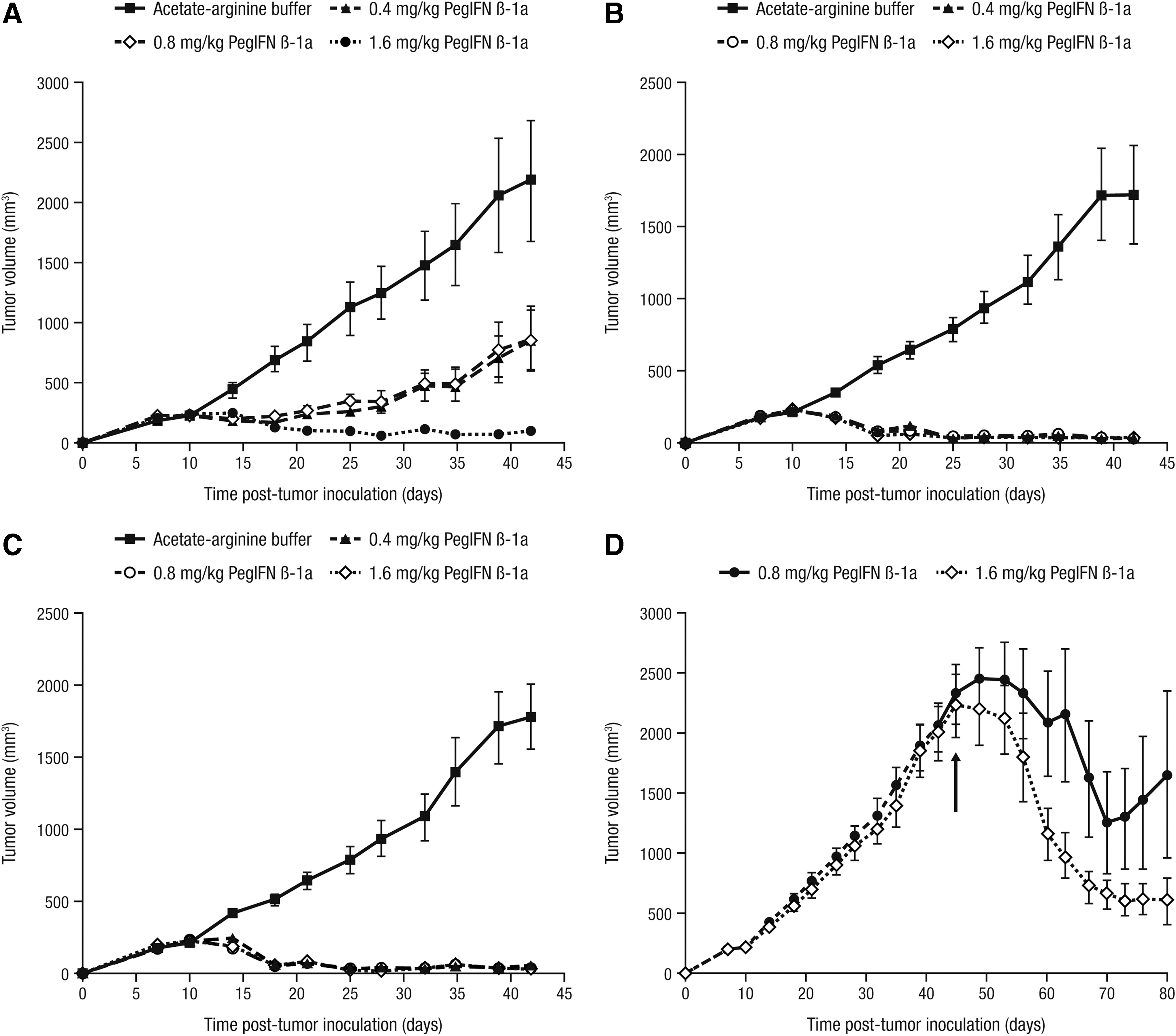
Peginterferon beta-1a induces regression of human WM-266-4 melanoma xenografts in nude mice. WM-266-4 cells were inoculated SC into the flanks of nude mice (n = 10), and tumors were staged to 200 mm3 before treatment with vehicle control (arginine–acetate buffer) or with 0.4, 0.8, or 1.6 mg/kg peginterferon beta-1a SC QW
Peginterferon beta-1a treatment of large WM-266-4 melanoma xenografts in nude mice
To determine whether peginterferon beta-1a would affect large established tumors, tumor regression was assessed in mice carrying WM-266-4 melanoma tumors (∼2000–2500 mm3) by treating with either 0.8 mg/kg (n = 9) or 1.6 mg/kg (n = 10) peginterferon beta-1a (from day 46 to day 74 post-tumor inoculation; Fig. 3D). While both doses regressed the large tumors, upon completion of 4 weeks of peginterferon beta-1a treatment (day 74 post tumor inoculation), tumors in mice treated with 0.8 mg/kg appeared to regrow, but the tumor regression induced by 1.6 mg/kg was maintained (Fig. 3D).
Inhibition of melanoma tumor growth by combination treatment with peginterferon beta-1a and the MEK kinase inhibitor PD325901
SK-MEL-1 melanoma tumor-bearing nude mice were treated with peginterferon beta-1a in combination with the MEK kinase inhibitor PD325901 to determine whether the growth inhibitory activity of peginterferon beta-1a could be augmented by concomitant treatment (Fig. 2A). Treatment with peginterferon beta-1a or PD325901 significantly inhibited SK-MEL-1 melanoma growth in nude mice (n = 10) compared with the vehicle (P < 0.0001). The dose of PD325901 combined with peginterferon beta-1a was chosen based on a preliminary study in which SK-MEL-1 tumor-bearing nude mice were treated with doses of 0 to 20 mg/kg PD325901, and in which the effect of 15 mg/kg was determined to be submaximal (data not shown).
Combination of peginterferon beta-1a and PD325901 significantly inhibited tumor growth compared with either treatment alone (P < 0.0001; Fig. 2A). Moreover, the difference between estimates of least squares means between the vehicle group and the treatment groups suggested an additive effect of peginterferon beta-1a and PD325901, that is, the value for the combination (−8.63) was approximately the sum of the values for peginterferon beta-1a (−4.77) and PD325901 (−4.52). Western blotting of extracts prepared from the tumors of mice treated with vehicle control, peginterferon beta-1a, PD325901, and peginterferon beta-1a + PD325901 showed that the phosphorylation of ERK, the substrate for MEK, was significantly inhibited in the presence of PD325901, confirming the on-target activity of the MEK inhibitor. By contrast, ERK was phosphorylated in the vehicle and peginterferon beta-1a-treated groups, indicating that the inhibition of tumor growth by peginterferon beta-1a was likely independent of the Ras/Raf/MEK/ERK pathway (data not shown).
Effect of combination treatment of peginterferon beta-1a and approved antitumor drugs in human breast, renal, and colon carcinoma xenograft models
In addition to studying its growth inhibitory effects in melanoma models, peginterferon beta-1a was also evaluated in combination with approved antitumor drugs to determine its activity in breast, renal, and colon cancer xenografts in nude or SCID mice.
In MDA-MB-468 breast carcinoma-bearing nude mice, 0.4 mg/kg (4 × 107 units/kg) of peginterferon beta-1a induced greater inhibition of tumor growth (n = 4) than 0.2 mg/kg (4 × 107 units/kg) of interferon beta-1a (P = 0.0256; Fig. 4A). In MDA-MB-231 breast carcinoma-bearing SCID mice, 0.4, 0.8, and 1.6 mg/kg of peginterferon beta-1a resulted in significant tumor growth inhibition compared with the vehicle control (P = 0.0008, P = 0.002, and P = 0.0004, respectively; Fig. 4B). Moreover, combination of peginterferon beta-1a at 0.8 or 1.6 mg/kg with 25 mg/kg paclitaxel was more effective in inhibiting and suppressing regrowth of the carcinomas than peginterferon beta-1a alone (P < 0.0001) or paclitaxel alone (P < 0.0001; Fig. 4C). Of note, 9 of the 10 tumors were barely palpable by day 77 and day 62 for the 0.8 mg/kg peginterferon beta-1a + paclitaxel and 1.6 mg/kg peginterferon beta-1a + paclitaxel groups, respectively, indicating that the combination treatments had, in essence, resulted in complete tumor regression.
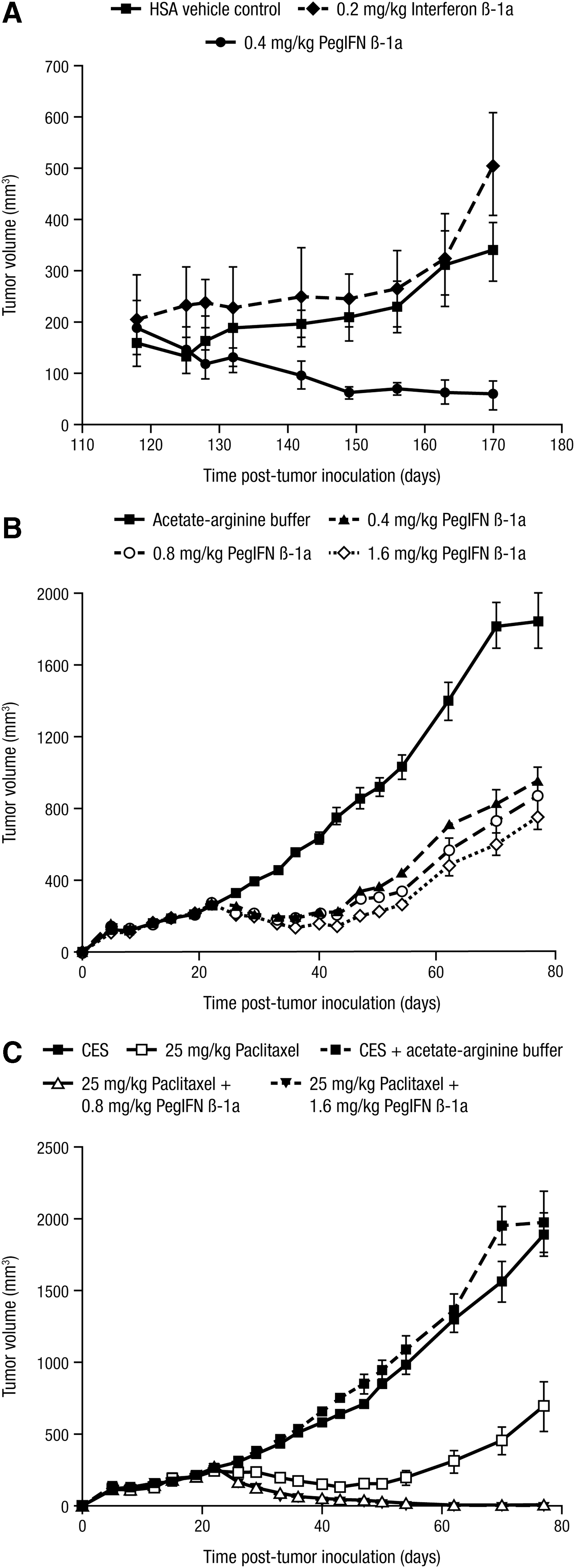
Peginterferon beta-1a suppresses tumor growth alone and in combination with paclitaxel in human breast carcinoma models.
To assess peginterferon beta-1a in a renal cell carcinoma model, SN12-C tumor-bearing SCID mice were treated with vehicle control (SC BIW for 7 weeks), peginterferon beta-1a (0.8 or 1.6 mg/kg SC BIW for 6 to 7 weeks), temsirolimus (50 mg/kg IP QW for 2 weeks), bevacizumab (4 mg/kg IP BIW for 7 weeks), or sorafenib [60 mg/kg per os (oral) (PO) once daily (QD) for 2 weeks], and combinations of peginterferon beta-1a with either temsirolimus, bevacizumab, or sorafenib (sorafenib dosed concurrently with peginterferon beta-1a and sequentially where the sorafenib was given first). SC treatment with 0.8 mg/kg peginterferon beta-1a alone inhibited tumor growth significantly compared with the vehicle control (P < 0.0001), with 1.6 mg/kg of peginterferon beta-1a significantly inhibiting tumor growth compared with vehicle (P < 0.0001) and 0.8 mg/kg peginterferon beta-1a (P = 0.0005; data not shown). Significant inhibition of tumor growth was observed with temsirolimus alone (P = 0.0285), but no significant differences were observed with bevacizumab alone (P = 0.1146) or with sorafenib alone (P = 0.5349) compared with the vehicle (data not shown).
For the combination of peginterferon beta-1a + temsirolimus in SN12-C tumor-bearing SCID mice, significant inhibition of tumor growth (n = 9–10) was observed compared with either agent alone (P < 0.0001; Fig. 5A). Similarly, combination of peginterferon beta-1a with bevacizumab led to significant inhibition of tumor growth in SCID mice (n = 9–10) compared with either agent alone (P < 0.0001; Fig. 5B). For the combinations of peginterferon beta-1a + sorafenib, significant inhibition of tumor growth in SCID mice (n = 9–10) was observed compared with either agent alone with concurrent dosing (P = 0.0007 versus peginterferon beta-1a alone and P < 0.0001 versus sorafenib alone) but, for sequential dosing, the difference was only significant versus sorafenib alone (P < 0.0001; Fig. 5C).
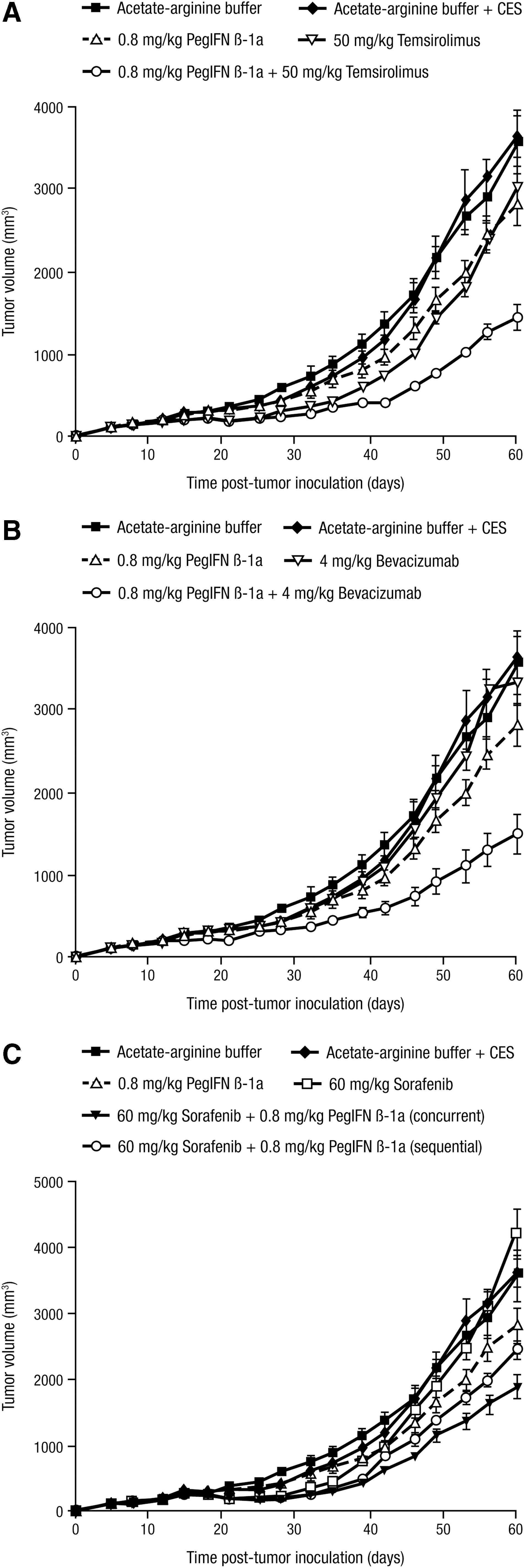
Peginterferon beta-1a suppresses tumor growth in combination with drugs approved for the treatment of renal cell carcinoma. SN12-C cells were inoculated SC in the flanks of SCID mice (n = 9–10), and tumors were staged to 200 mm3 before treatment with
In SW-620 human colon carcinoma tumor-bearing nude mice, 0.4, 0.8, or 1.6 mg/kg peginterferon beta-1a was ineffective at inhibiting tumor growth (n = 9–10) compared with the vehicle control. Statistical analysis of the data indicated that no significant differences existed between the four treatment groups (P = 0.312 for the treatment by day interaction), so no contrasts for comparisons were warranted; see Figure 6 for the 0.8 mg/kg dose group. By contrast, both irinotecan (10 mg/kg) and bevacizumab (4 mg/kg), agents approved for the treatment of colon cancer in humans, had significant antitumor activity compared with the vehicle control (P < 0.0001), with the antitumor effect of irinotecan being significantly greater than that of bevacizumab (P < 0.0001). While significantly greater tumor growth inhibition was achieved with 0.8 mg/kg peginterferon beta-1a + 4 mg/kg bevacizumab compared with either drug alone (P < 0.0001), the antitumor activity of irinotecan did not increase when combined with peginterferon beta-1a (P = 0.0793).
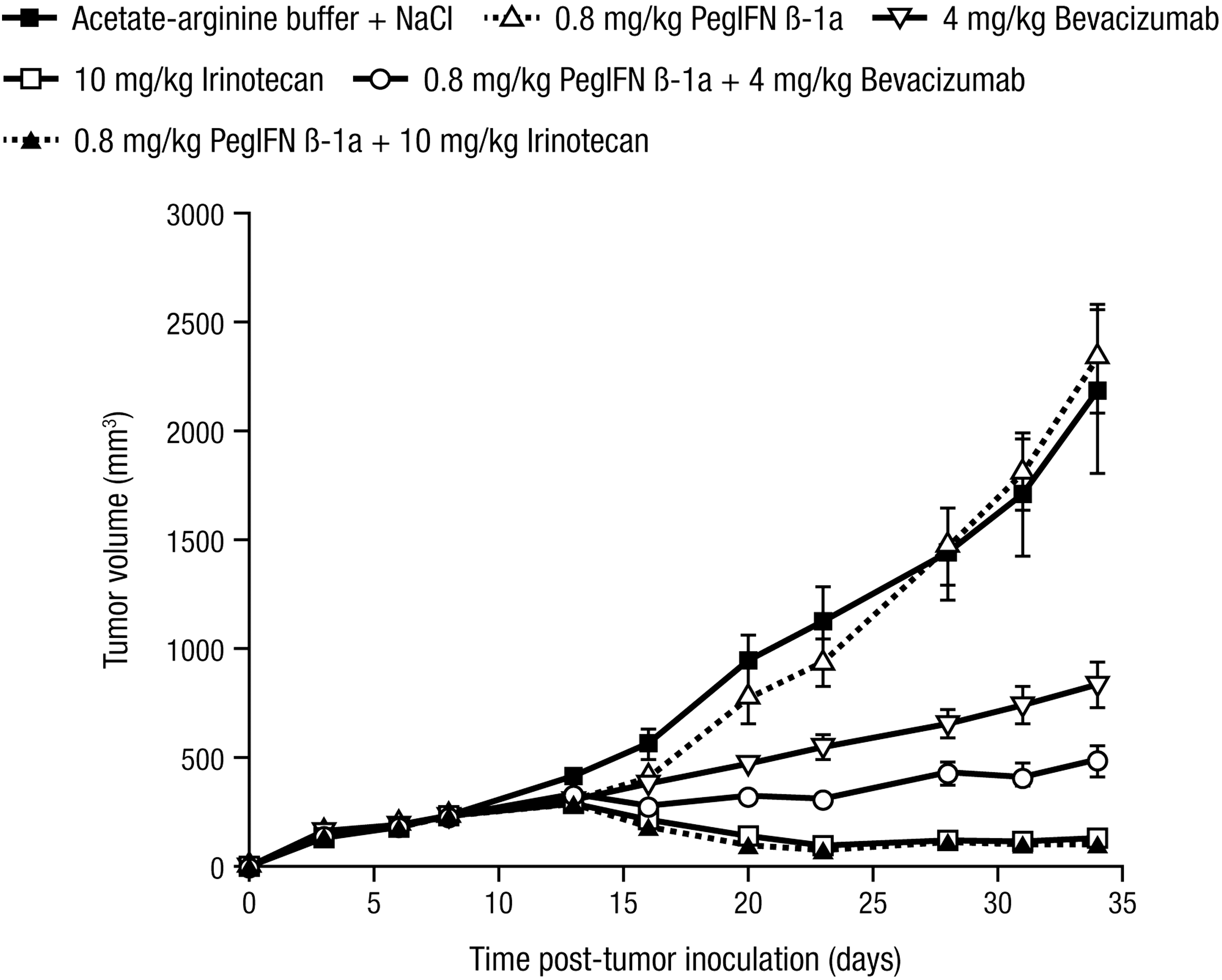
Peginterferon beta-1a suppresses tumor growth in combination with drugs approved for the treatment of colon carcinoma. SW-620 cells were inoculated SC in the flanks of nude mice (n = 10), and tumors were staged to 200 mm3 before treatment with vehicle control (acetate–arginine buffer) SC BIW for 4 weeks, 0.8 mg/kg peginterferon beta-1a SC BIW for 4 weeks, 4 mg/kg bevacizumab IP BIW for 4 weeks, 10 mg/kg irinotecan PO QD for 10 days, 0.8 mg/kg peginterferon beta-1a SC BIW for 4 weeks + 4 mg/kg bevacizumab IP BIW for 4 weeks, and 0.8 mg/kg peginterferon beta-1a SC BIW for 4 weeks + 10 mg/kg irinotecan PO QD for 10 days. Error bars depict standard error. BIW, twice weekly; IP, intraperitoneal; PO, per os (oral); QD; once daily; SC, subcutaneous.
Effect of treatments on mouse weight
For all agents tested, except for sorafenib, mouse weight was either maintained or increased during the studies. In mice treated with sorafenib, a significant weight loss occurred during the dosing period. Weight loss was transient with mice typically losing ∼14% of their body mass. However, following cessation of treatment, they regained the lost weight and continued to grow.
Discussion
Pegylation is an established methodology that has been utilized to improve the PK properties of therapeutic proteins (Kozlowski and others 2001; Harris and Chess 2003). The benefits of this method, as demonstrated by the pegylated forms of interferon alpha for the treatment of hepatitis C virus infection (Glue and others 2000; Rajender and others 2002), are less frequent dosing and enhanced clinical efficacy. Similar benefits have been noted with pegylated G-CSF for the treatment of chemotherapy-induced neutropenia and pegylated interferon beta-1a for the treatment of relapsing remitting multiple sclerosis, both of which retained the efficacy of their respective unmodified forms with a reduced dosing schedule (Crawford 2002; Calabresi and others 2014).
In the present study, peginterferon beta-1a induced cell death of WM-266-4, SK-MEL-2, SK-MEL-5, and MeWo cells following in vitro treatment. In contrast, SK-MEL-1 cells were resistant to peginterferon beta-1a-induced cell death in vitro, but, conversely, inhibition of tumor-induced angiogenesis (Baker and others 2006) and growth of SK-MEL-1 melanoma tumors in mice was inhibited by peginterferon beta-1a. This suggests that the anti-SK-MEL-1 tumor activity of peginterferon beta-1a in mice may be mediated through the inhibition of angiogenesis, itself stimulated by tumor-expressed proangiogenic factors such as VEGF the expression of which is known to be inhibited by interferon beta (Yildirim and others 2015), rather than through a direct antiproliferative effect on the tumor cells. This characteristic of SK-MEL-1 cells has been observed previously when human interferon alpha-2b and thalidomide inhibited tumor-induced angiogenesis and tumor growth in nude mice, but did not induce antiproliferative effects in vitro (Bauer and others 2003).
Since WM-266-4 cells were the most sensitive to peginterferon beta-1a treatment in vitro, they were chosen for analysis of the apoptotic pathways involving activation of TRAIL and PARP, as well as cleavage of caspases 3, 8, and 9 in response to treatment. Robust peginterferon beta-1a-induced cell death was observed following treatment of WM-266-4 cells. A similar apoptotic mechanism involving activation of TRAIL and cleavage of caspases 3 and 8 was reported previously in breast cancer cells treated with retinoic acid and the toll-like receptor (TLR) agonist poly (I:C) (Bernardo and others 2013).
The results of the current study demonstrate the effectiveness of peginterferon beta-1a monotherapy in inhibiting the growth of human melanoma tumors in murine xenograft models. Although peginterferon beta-1a alone was effective in inhibiting melanoma growth, studies have shown that solid tumors are generally resistant to treatment with unmodified interferons (Willimsky and others 2005). Therefore, peginterferon beta-1a treatment in combination with the MEK kinase inhibitor PD325901 was assessed to determine its effectiveness compared with each monotherapy. Combination of peginterferon beta-1a and PD325901 significantly inhibited SK-MEL-1 melanoma tumor growth compared with treatment with either agent alone, and did so in a manner suggestive of an additive effect.
As the Ras-Raf-MEK-ERK pathway is susceptible to mutations that may drive melanomas, the effect of combining peginterferon beta-1a with small molecule inhibitors directed against other kinases of the pathway will be an important avenue of future research. However, it has been reported (Suresh Kumar and others 2007) that B-Raf inhibition using sorafenib suppresses Jak-Stat signaling in B-Raf-activated melanoma cells, leading to the attenuation of the antiproliferative activity of interferon alpha. If this in vitro observation translates to the in vivo setting, it is possible that the combination of peginterferon beta-1a with sorafenib (or potentially any B-Raf inhibitor) may not result in additive or synergistic effects in melanoma tumors, even though the combination of peginterferon beta-1a + sorafenib resulted in significant inhibition of tumor growth compared with either agent alone in SN12-C renal cell carcinomas grown in SCID mice (Fig. 5C).
In addition to inhibition of melanoma tumor growth, peginterferon beta-1a improved the effectiveness of anticancer drugs approved for the treatment of renal, breast, and colon carcinomas. Similar to our findings, a previous study demonstrated that a combination of interferon beta and tamoxifen increased the inhibition of breast tumor cell growth compared with either agent alone in vivo and in vitro (Lindner and Borden 1997). However, while combination treatment of peginterferon beta-1a with bevacizumab suppressed colon carcinoma growth, and did so in an apparent synergistic manner since peginterferon beta-1a alone was ineffective but increased the effectiveness of bevacizumab, combination of peginterferon beta-1a with irinotecan failed to augment its antitumor activity in the colon xenograft tumors. The lack of an additive or synergistic effect in the latter combination may be explained, in part, by the strong inhibition of tumor growth imparted by irinotecan alone, that is, tumor growth inhibition using 10 mg/kg irinotecan PO QD for 10 days may have been saturating and a lower concentration may be required to show an additive or synergistic effect in combination with peginterferon beta-1a.
While interferon alpha treatment in combination with 5-fluorouracil failed to provide a survival benefit and was associated with increased toxicity in patients with stage III colon cancer in a prospective clinical trial (Schippinger and others 2005), the use of pegylated interferon beta-1a in such a patient population has yet to be determined. A summary of the antitumor effects of peginterferon beta-1a, alone or in combination with the various anticancer drugs tested, is shown in Table 1.
+, denotes inhibition of tumor growth; −, denotes no inhibition of tumor growth; ++, denotes inhibition greater than either single agent alone; NT, not tested.
A limitation of studying the effect of peginterferon beta-1a in murine xenograft models is that due to the species specificity of human and murine interferon beta (Qin and others 2001; Arduini and others 2004), and that nude and SCID mice are immune compromised, the potential role of the immune system in tumor growth inhibition and/or regression in humans cannot readily be addressed. Indeed, the contribution of the immune system in interferon beta-mediated tumor growth inhibition is nicely summarized in studies of human and murine interferon β-expressing replication-deficient adenovirus in nude mice xenografts (Qin and others 2001). In these studies, the effect of treating human MDA-MB-468 breast carcinoma and human PC-3 prostate xenografts in nude mice was evaluated. While both human and murine interferon β-expressing adenovirus resulted in complete tumor regression and animal survival when the vector was injected into the tumors, tumor regression was significantly faster with delivery of human interferon beta than with murine interferon beta.
Being that: (1) murine interferon beta protein showed no antiproliferative activity against human MDA-MB-468 breast carcinoma cells in vitro even at 30,000 units/mL (by contrast, human interferon beta-1a protein showed significant inhibition at 100 units/mL); (2) depletion of NK cells in nude mice had no effect on tumor regression or animal survival following delivery of human interferon beta, but significantly inhibited tumor regression and animal survival following delivery of murine interferon beta; and (3) tumors in both left and right flanks regressed when only the right side tumor was treated with murine interferon β-expressing adenovirus, the role of interferon beta in mediating immune system tumor regression is clear. Moreover, in syngeneic immunocompetent mice, tumor regression and animal survival following delivery of murine interferon β-expressing adenovirus was mediated by a CD8+ T-cell immunologic antitumor response (Brown and others 2002).
Therefore, while the high doses of peginterferon beta-1a used in this study (0.1–1.6 mg/kg) are unlikely to be tolerated in cancer patients due to potential toxicities (in contrast, the approved dose of peginterferon beta-1a (Plegridy®) for the treatment of relapsing remitting multiple sclerosis is 125 μg once every 2 weeks, independent of patient weight (Plegridy® prescribing information 2014), the role of the immune system in peginterferon beta-1a-mediated antitumor effects in humans may enable lower doses to be used. Since peginterferon beta-1a is unlikely to be used as a monotherapy for the treatment of cancers, potential additive or synergistic effects when combined with standard of care therapies may also permit lower doses to be used.
In conclusion, these findings suggest that peginterferon beta-1a may have potential as a treatment option for melanoma, as well as for solid tumors such as breast, kidney, and colon cancers.
Footnotes
Acknowledgments
The study was sponsored by Biogen, Inc. (Cambridge, MA, USA). The authors were assisted in the preparation of the article by Asha Jayakumar of Biogen, Inc., and by Jason Jung of MedErgy. Writing support was funded by the study sponsor.
Author Disclosure Statement
All authors declare no conflicts of interest with the exception of Margot Brickelmaier and Xiao Hu, who are employees of Biogen, Inc. All other authors are former employees of Biogen, Inc. with the exception of Daniel Lindner and Thomas Scripps.