
Abstract
The interferons (IFNs) are a family of cytokines with diverse cellular actions such as control of cell proliferation and regulation of immune responses; therefore, they have been extensively studied as antitumor agents for a variety of malignancies, including gliomas. Type I IFNs exert their antitumor effects either directly, by targeting the tumor cells or the tumor stem cells, or indirectly, by regulating the anticancer activities of the immune system. More specifically, IFN-beta and IFN-alpha exhibit antiproliferative effects by p53 induction, CD8+ T-lymphocyte and macrophage activation, chemokine secretion, and miR-21 downregulation. In vitro and in vivo studies provide evidence that immunotherapy could have a role in glioma treatment, especially when first-line therapeutic interventions fail to produce durable responses. These effects are more obvious when combining IFN-beta with classical antitumor therapies such as temozolamide, an oral chemotherapeutic, for both newly diagnosed and recurrent gliomas. However, further clinical studies are needed to determine whether IFNs will have a definite place in the management of gliomas.
Introduction
T
IFNs exert their antitumor effects by inducing gene transcription and/or multiple downstream signaling that both inhibit the development of glioma cells in S-phase, induce apoptosis, enhance natural killer (NK) cell and cytotoxic T cell activity, and synergize with chemotherapeutic agents (Marras and others 2003; Galani and others 2016).
Another regulator signaling involved in IFN-mediated inhibition of glioma growth is the increase of macrophage cytotoxicity toward glioma cells. Kito and others reported that macrophage activation is associated with generation of nitric oxide synthase (iNOS) and nitric oxide (NO) (Kito and others 2002). In addition, laboratory animals bearing cerebral gliomas immunized with IFN-gamma-releasing N32 glioma cells display significant increase in iNOS expression at the immunization locus and in brain tumors (Johansson and others 2002). However, due to the complexity of IFN signaling, the full role of IFNs on the tumor cell death and invasion is not completely understood.
The aim of this review is to summarize the biological results of type I IFNs in gliomas and correlate them with their potential clinical significance.
Type I IFNs signaling mechanisms
Type I IFNs initiate their signaling by binding to the type I IFN receptor (IFNAR), which is constituted of 2 subunits, IFNAR1 and 2. Binding of a type I IFN to the receptor triggers heterodimerization of IFN-alpha/beta receptor subunits and initiates a sequence of procedures leading to transcriptional activation of IFN-stimulated genes that induce biologic response. To start with, the Janus activated kinase–signal transducer and activation of transcription (JAK-STAT) pathway can directly impart signals from the cell surface to the nucleus. IFNAR1 is related to tyrosine kinase 2 (TYK2), while IFNAR2 is linked to JAK1. Receptor heterodimerization leads to JAK1 and TYK2 transphosphorylation and, consequently, to phosphorylation of the cytoplasmic part of the IFNAR in its tyrosine phosphorylation sites. These phosphotyrosyl residues will be the docking for the STAT proteins and will provoke their phosphorylation and dimerization. Once dimerized, the STATs translocate to the nucleus where they bind to the protein p48 (also called IFN-regulatory factor 9 [IRF9]) and induce the formation of the Interferon Stimulated Gene Factor 3 (ISGF3) which binds to IFN-Stimulated Response Elements (Darnell 1997; Uddin and others 1999). Type I IFNs activate mainly STAT1, STAT2, and STAT3, STAT5 is also found to be activated, which may cause diverse cellular responses. Type I IFNs mainly induce activation of STAT1 and STAT2; STAT3 and STAT5 are found to be activated as well, which may cause the diverse cellular responses. The activation of JAK–STAT pathway also leads to accumulation of myxovirus resistance 1 (MxA) protein, a GTPase interfering with the cytoskeletal structure (Haller and Kochs 2002). MxA induction is a recognized marker for responsiveness to type I IFNs (von Wussow and others 1990; Holzinger and others 2007). Activation of the IFNAR/JAK/STAT1 pathway can also cause the upregulation of the TAM receptor Tyrosine Kinase Axl, which can hijack IFNAR-induced STAT1 activation through suppressors of cytokine signaling (SOCS) proteins (Alexander 2002).
Besides, the classical JAK/STAT signaling pathway, there are alternative pathways that are responsible for the responses to type I IFNs, namely the v-CRK avian sarcoma virus CT10 homolog-like (CRKL) pathway, the mitogen-activated protein kinase (MAPK) pathway, the phosphoinositide 3-kinase (PI3K) pathway, and either the conventional or the unconventional NF-κB cascade (Platanias 2005).
Type I IFNs induce tyrosine phosphorylation of CrkL, a protein with Src Homology 2 and 3 (SH2 and SH3) domains, resulting in the formation of a signaling complex with Stat5 that moves to the nucleus and binds specific GAS elements. (Yongzhong and others 2004). With respect to MAPK signaling, activation of JAK causes the phosphorylation of the guanine nucleotide exchange factor Vav, which results in activation of Rat sarcoma protein (Ras) and Ras-related C3 botulinum toxin substrate 1 (Rac1). This consequently may lead to activation of many proteins belonging to MAPK family, namely p38, c-Jun N-terminal kinases (JNK), and extracellular signal-regulated kinases (ERK) 1 and 2. The diverse MAPKs induce the activation of diverse transcription factors and are responsible for the variety of IFN-I effects on immune system. The PI3K/Akt is a key signaling pathway, as well, since PI3K has both serine and lipid kinase action. Activation of PI3-kinase induces the release of the intracellular phosphorylated lipid such as phosphatidylinositol-3,4-P2 and phosphatidylinositol-3,4,5-P3, which act as second messengers. PI3K induces downstream signaling such as the serine–threonine kinase Akt that induces downstream activation of mTOR and the inactivation of numerous proteins, including glycogen synthase kinase 3 (GSK-3) and cyclin-dependent kinase inhibitors 1A and 1B (CDKN1A and CDKN1B) which are implicated in cell division regulation. Finally, the NF-kappaB pathway can be activated either by the AKT pathway by the IκB kinase beta (IKKβ) or by binding of TNF receptor-associated factors (TRAF) with the activated NF-κB-inducing kinase (NIK), which successively leads to activation of NF-kappaB by the IκB kinase alpha (IKKα) (Hoesel and Schmid 2013).
Antitumor activity of type I IFNs
Type I IFNs cause diverse cellular responses, such as differentiation, growth arrest, and apoptosis. The antitumor effect of type I IFNs is direct by inducing growth arrest and apoptosis of tumor cells and indirect by activation of the host immune response (Galani and others 2016) (Table 1). More precisely, expression of histocompatibility complex class I (MHC I) is upregulated by type I IFNs resulting in the enhancement of antigen recognition. In addition, they provoke the expression of adhesion molecules such as ICAM-1 and L-selectin (Martín-Henao and others 2000). It has also been shown that IFN alpha suspends the secretion of tumor-derived proangiogenic factors such as b-fibroblast growth factor (bFGF) (Slaton and others 1999). Furthermore, it is now known that type I IFNs stimulate the secretion of IL-15, which lead to proliferation of NK cells and CD8 T cells (Nguyen and others 2002). There is also evidence that toll-like receptor (TLR)-stimulated cross-presentation by classical dendritic cells (cDCs) induces the antitumor immunity, and STAT2 causes the TLR-induced dendritic cell stimulation and cross-presentation (Xu and others 2016).
The prodeath effect of type I IFNs is principally mediated by the transcriptional regulation of proapoptotic genes such as ISGs that are activated by the death ligand TNF-related apoptosis-inducing ligand (TRAIL) (Chawla-Sarkar and others 2003; Borden and others 2007).The same way, IFN-beta promoter stimulator-1 (IPS-1) induces anticancer activity either through TRAIL upregulation or through downregulation of the proteins BCL2, BIRC3, and PRKCE that inhibit apoptosis in IFN I-dependent or IFN I-independent way (Kumar and others 2015).
However, cancer cells often acquire resistance to IFNs mediated by alterations in the IFN signaling pathway. Deficiencies in IRF-9 and Stat2 expression or epigenetic alterations cause resistance to type I IFN-mediated antitumor effect (Reu and others 2006; Du and others 2009; Romero-Weaver and others 2010). Upregulation of the antiapoptotic Bcl-2 family members and activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway can also cause resistance to type I IFN-induced cell death (Lesinski and others 2008; Sheng and others 2010). Furthermore, SOCS 1 is a negative regulator of type I IFN signaling pathways through an interaction with the TYK2 that is part of the IFNAR1 (Piganis and others 2011) and, therefore, SOCS1 silencing induces antitumor effect of Type I IFNs through the modulation of apoptosis in different cells (Zitzmann and others 2007; Lesinski and others 2010).
Type I IFNs regulate more than 2,000 coding and noncoding RNA transcripts, called IFN-regulated genes (IRGs). Mi-RNAs seem to play a pivotal role in modulating the immune response related to IFNs; they are negative factors of activation by targeting signaling molecules or increase signaling through repression of negative modulators (Samuel and others 2015). MiRNA-145,211, and 222 downregulate the expression of STAT-1 protein, which is associated with the signaling through any of the IFN receptor complexes (Gregersen and others 2010; Kohanbash and Okada 2012), and miR-221 and miR-222 decrease gene and protein expression of STAT1 and STAT2, which are induced by IFN-alpha (Zhang and others 2010) (Fig. 1).
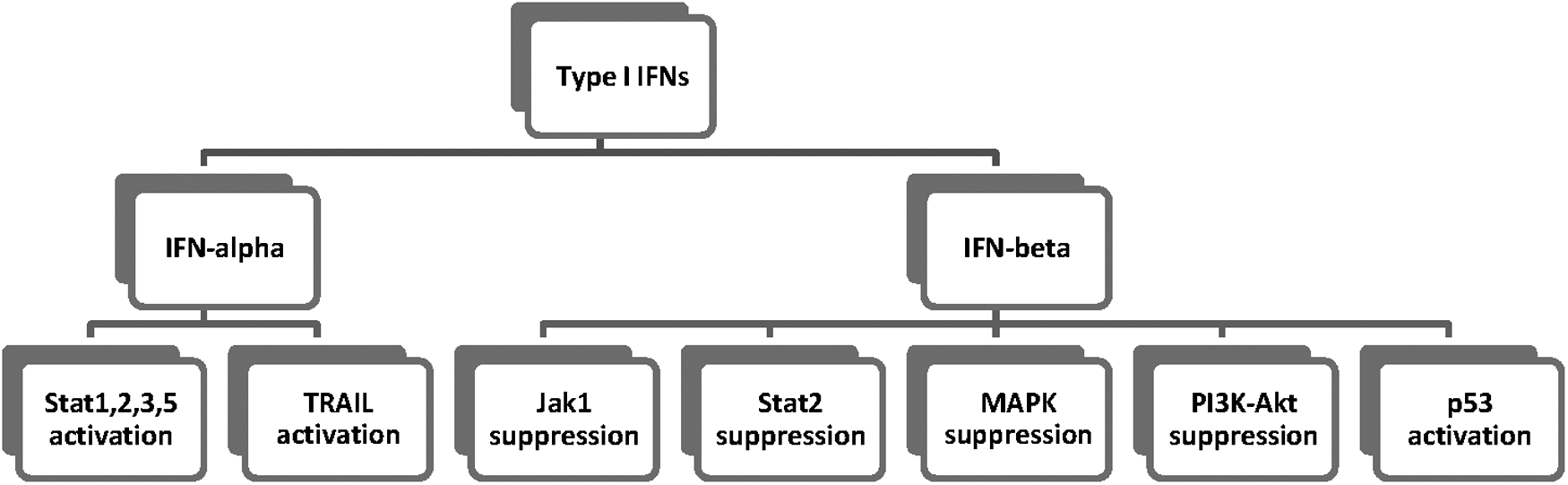
The major pathways mediated by Type I IFNs. JAK, Janus kinase; MAPK, mitogen-activated protein kinases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; STAT, signal transducers and activators of the transcription family of proteins; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B (PKB); TRAIL, TNF-related apoptosis-inducing ligand.
In addition, IFN signaling may induce or suppress directly the microRNAs. To start with, microRNAs regulate type I IFN signal transduction through SOCS1; miR19a, miR122, and miR155 have been shown to be associated with SOCS1 (Pichiorri and others 2008; Jiang and others 2010; Li and others 2013). All in all, the modulation of apoptotic signaling induced by type I IFNs could reduce resistance of glioma cells to antitumor agents.
Effects of type I IFNs on gliomas (in vitro and in vivo studies)
IFNs were put into practice in the treatment of solid malignancies early in 1970 s, without being combined with radiotherapy and/or chemotherapy (Nagai and Arai 1984). When referring to glioblastoma, a number of studies in tissue cultures (Lundblad and Lundgren 1981; Cook and others 1983; Yates and others 1985; Strayer and others 1987) as well as in nude mice had been initially performed. Cook and others and Tanaka and others indicated that IFNs inhibit the development of neoplastic cells conserved in tissue culture and transplanted into nude mice (Cook and others 1983; Tanaka and others 1983). Accordingly, Dick and others studied the possible antiproliferetive effects of IFNs as well as double-stranded RNAs (dsRNAs) on 5 human glioma cell lines, A172, A1207, A1235, A1690, and A2781. They confirmed the antiproliferative effects of IFNs individually and synergistically with combinations of type I and type II IFNs. In addition, the IFN-alpha/IFN-beta combination gave an additional antitumor result (Dick and Hubbell 1987). In these studies, IFN-beta seemed to have a better antitumor result compared to IFN-alpha, yet the molecular events remained unclear.
It is now well established that type I IFN-mediated signaling increases IFN-alpha/beta stimulation in response to virus, through autocrine/paracrine upregulation of IRF-7. (Hata and others 2001). The constitutive IFN signaling can also impede oncogenic transformation and it is abolished in unprompted cellular transformation (Chen and others, 2009; Kulaeva and others, 2003). Thus, the biological effects of type I IFNs are correlated with low levels of expression.
Indeed, U87MG cells, which hold a deletion of type I IFN genes and T98G cells, were treated with 1,000 U/mL of IFN-alpha. Both glioma cell lines were found to be resistant to IFN-alpha-induced apoptosis even with the presence or absence of IFN signaling pathway; downregulation of ubiquitin-specific peptidase USP18 sensitized T98G, but not U87MG cells to IFN-alpha-induced apoptosis. Endoplasmic reticulum (ER) stress sensitized U87MG cells to apoptosis induced by TRAIL and TRAIL mRNA stability was found to be under IFN-alpha control (Fig. 1). The downregulation of USP18 expression and the induction of ER stress are involved in restoration of apoptosis in U87MG cells. In addition, it was suggested that unprompted IFN signaling, probably through the regulation of IRF-1 levels, could conduce to maintain TRAIL expression in glioblastoma cells (Sgorbissa and others 2011).
Stat-3 has also been shown to be associated with antiproliferative functions when activated by IFN-alpha/beta in human Daudi cells (Homo sapiens, human peripheral blood, B lymphoblast, cell lines) (Yang and others 1998); in glioma cells, this may result in IFN-beta-mediated tumor inhibition through negative regulation of miR-21 (Ohno and others 2009). Consequently, miR-21 has been indicated to be overexpressed in numerous tumors and may be a possible clinical biomarker with therapeutic implications (Chan and others 2005). Significantly increased miR-21 levels have been observed in glioblastoma (Hermansen and others 2013). In addition, Galani and others (2015) reported that atypical and malignant meningiomas had higher miR-21 levels than benign meningioma.
On the other hand, IFN-beta has been widely used in in vitro and in vivo studies. IFN-beta induces diverse biological responses, including cellular differentiation, cell development, and immune responses. IFN-beta is also a cell cycle regulator, causing altered cell cycle development, which may occur as S phase cumulation, and less often as G1 arrest, or entrance of tumor cells into a senescence-like status (Kaynor and others 2002).
There is evidence that homozygous deletions of the type I IFN gene cluster, including several IFN-alpha genes and a single IFN-beta gene, occur in human glioma and melanoma. Thus, human IFN-beta plays a pivotal role in the modulation of human glioma and melanoma growth (Miyakoshi and others 1990; James and others 1991).
MiRNAs in tumors may act either as oncogenes or as tumor suppressors (Calin and others 2004). Tanaka and others treated TE671, ONS-76 human medulloblastoma cell lines, and A172, U251-MG glioblastoma cell lines with recombinant human IFN-beta and found that viability of medulloblastoma and glioblastoma cell lines was repressed in a dose- and time-dependent manner. It is suggested that IFN-beta suppressed Jak1 and Stat2 by upregulation of suppressor of cytokine signaling 6 (SOCS6), a cytokine-induced Stat inhibitor, in both medulloblastoma and glioblastoma cells (Fig. 1). In addition, mitogen-activated protein kinase (MAPK) was suppressed only in medulloblastoma cells and phosphatidylinositol-3 kinase (PI3K-Akt) only in glioblastoma cells (Fig. 1). Yet, cell development was not repressed after 48-h treatment with human IFN-beta and impermanent transfection with miR-431, as SOCS6 was found to be downregulated (Tanaka and others 2014). Thus, combination therapy with downregulation of miR-431 and a PI3K-Akt or MAPK inhibitor might probably generate better therapeutic effects to IFN-beta-resistant brain tumors.
Moreover, the combination of IFN-beta with Temozolamide (TMZ), a first-line oral chemotherapeutic agent, has been adequately studied. Natsume and others (2005) treated in vitro human glioma cell lines (T98, AO2, SKMG1, U251nu/nu, U251SP, and U251MG) with IFN-beta and TMZ; their study indicated that IFN-beta sensitized gliomas with the unmethylated O6-methylguanine DNA methyltransferase (MGMT) promoter, which are resistant to TMZ, possibly because of reduction of MGMT expression by p53 stimulation. In the same context, Natsume and others used an animal model with T98 (TMZ-resistant) and U251SP (TMZ-sensitive) tumors and treated the objects with animal IFN-beta and TMZ. They found that combined therapy significantly decreased tumor growth in TMZ-resistant tumors as IFN-beta enhanced p53-mediated MGMT inhibition and sensitized glioblastoma cells to TMZ (Natsume and others 2008) (Fig. 1).
Yoshino and others (2009) came to similar conclusions when they cotreated in vitro human malignant glioma A-172, AM-38, T98G, U-251 MG, YH-13, U-87 MG, and U-138 MG cells with IFN-beta and TMZ, and additionally suggested that TMZ is responsible for endogenous IFN-beta stimulation in T98G cells and other cell lines in a time-related manner, and therefore, TMZ was found to be an IFN inducer. Thus, the combination of IFN-beta and TMZ chemotherapy increases tumor sensitivity to TMZ. Further studies are needed to define the complete role of combined IFN-beta and TMZ chemotherapy to increase tumor sensibility and minimize tissue damage and potential toxicities. The mechanism that is accountable for the generation of endogenous IFN-beta is not clearly understood, but it seems that endogenous IFN-beta has a stronger antiproliferative effect on malignant gliomas compared to exogenous one (Mizuno and Yoshida 1998; Natsume and others 1999, 2000; Osawa and others 2005). It is suggested that endogenously produced IFN-beta induces the expression of Programmed death-ligand 1 (PD-L1) in glioma cells, tumor-infiltrating lymphocytes, and neurons surrounding glioma tissue; and the latter appears to be linked to prolonged survival in patients with malignant glioma (Nduom and others 2016).
In another experimental glioma study, Ito and others (2010) examined whether intravenously administered human neural stem cells (NSCs), which express cytokine diamenase (CD) and IFN-beta, immigrate into the intracranial tumor bed and if they exhibit antitumor effect. In the same in vivo and in vitro experiments, they used 5-fluorocytosine (5-FC) as cotreatment. They concluded that, in vitro F3.CD.IFN-beta human NSCs producing human IFN-beta demonstrate higher bystander killing result on glioma cells (Ito and others 2010). In addition, they claim that the clonogenic potential of the U251 cell survival after cotreatment with F3.CD.IFN-beta and 5-FC treatment was considerably lower than the clonogenic potential for the same U251 cells that were cocultured with F3.CD cells. In vivo results are similarly interesting; F3.CD.IFN-beta cells had the similar migrant pattern as that of the parental F3 cells. Moreover, the remaining tumor mass in the group treated with F3.CD.IFN-beta was lesser than that received by the group treated with F3.CD cells. Finally, the mice treated with F3.CD.IFN-beta survived considerably longer than the one treated with F3.CD or F3 cells (Ito and others 2010). Therefore, multimodal NSC-based treatment could be a potentially promising therapy for gliomas.
Apart from experimental studies, there is clinical evidence that IFN-beta associated with glioma treatment. Motomura and others (2011) retrospectively studied 68 patients with gliomas; 29 of them received radiotherapy and TMZ and 39 of them additionally received 3 × 106 IU/body of IFN-beta i.v. The overall survival was higher for the patients who received the combined therapy contrasted to the TMZ-alone group, and the benefit was especially more prominent in patients with an unmethylated MGMT promoter (Motomura and others 2011). Consequently, IFN-beta enhanced chemosensitivity to TMZ by downregulation of MGMT transcription. A multicenter phase 1 trial of IFN-beta/TMZ combination therapy for newly diagnosed malignant gliomas (INTEGRA study) was accomplished in Japan and revealed a protracted median overall survival with good tolerability (Wakabayashi and others 2011). More recently, Kawaji and others (2015) added IFN-beta to the adjuvant TMZ monotherapy in 7 patients with uncontrollable malignant glioma. Imaging findings and clinical symptoms were improved in large number of the cases, resulting in prolonged overall survival (Kawaji and others 2015).
A report of a case of a patient with recurrent glioblastoma and chemotherapy-induced myelosuppression by Utsuki and others (2011) suggested that even IFN-beta monotherapy may be a treatment option as they managed to control the tumor size with 600 × 104 IU of IFN-beta, 3 times a week for 6 weeks. Furthermore, a pilot clinical trial by Yoshida and others with 5 glioma patients who were treated with IFN-beta gene therapy after continuing progressing with previous surgery, radiotherapy, and chemotherapy showed stabilization or improvement of the general neurologic condition of 4 out of 5 patients. Subsequent pathologic examination of their tumors showed that gene therapy had direct antitumor results, namely apoptosis and tumor necrosis, as well as indirect antitumor results that are stimulation of immune cells, specifically of CD8+ T- lymphocytes and macrophages (Yoshida and others 2004). A phase I clinical trial with liposomal distribution of IFN-beta demonstrated small toxicity and clinical response with more than 50% decrease of the tumor in 2 out of 5 patients; histological examinations revealed numerous necrotic malignant cells, CD8+ T-Lymphocyte and macrophage infiltration, and decrease of CD34-immunoreactive vessels in the vector-injected area (Wakabayashi and others 2008). This study was followed by another phase I trial using administration of a human IFN-beta (hIFN-beta)-expressing adenovirus vector (Ad.hIFN-beta) into the tumor bed and the nearby tissue before and after surgical deduction of tumor, indicating protection and inductance of tumor cell apoptosis (Chiocca and others 2008).
Finally, there is accumulated evidence that IFN-beta gene transportation could stimulate apoptosis in interferon-beta protein-resistant tumor cells, including glioma, melanoma, and renal cell carcinoma. Glioma cells transduced by the IFN-beta gene produce interleukin (IL)-1β, IL-6 (IL-6 correlates with neoplasia) (Galani and others 2002), tumor necrosis factor (TNF)-α, monocyte chemotactic protein (MCP)-1, IFN-gamma-inducible protein-10 (IP-10), and heat shock protein (HSP), in addition to IFN-beta (Yoshida and others 2004). Therefore, the mix of these cytokines and the induction of apoptosis cause a potent antitumor effect on glioma cells. Such effects have also been noticed in other tumor cells such as lung carcinoma and prostate carcinoma, and have been attributed to several mechanisms (Galani and others 2009, 2010; Kastamoulas and others 2013; Chondrogiannis and others 2014). In summary, the inhibition of tumor development through cytokine signaling is cytokine specific and tumor cell-type specific.
Conclusion
The IFN signaling includes complicated mechanisms. The activity of IFNs is mediated through their receptors inducing transcription of genes and activation of immune responses that both lead to glioma cell growth inhibition and regulation of tumor invasion. In general, most immunotherapeutic strategies for gliomas include therapy with interferons. Interferons have a potent immunomodulatory capacity and their use is relatively safe with variable efficacy. Therefore, the regulation of IFN antitumor potency is IFN-type, cell-type, and cytokine combination specific. Such therapies may reduce resistance of glioma cells to various antitumor agents and could be valuable adjunct to chemotherapy.
Footnotes
Author Disclosure Statement
No competing financial interests exist.