
Abstract
Aicardi–Goutières syndrome (AGS) is an early-onset, genetic disease characterized by recurrent fever, multifocal lesions of the brain, and systemic autoimmunity. We report on 3 AGS patients, 2 siblings with an RNASEH2A gene mutation and 1 patient with a SAMHD1 gene mutation. Serial analysis of peripheral blood from all 3 AGS patients showed consistently elevated expression of the interferon-stimulated genes (ISGs): ISG15, RSAD2, and IFI27, not observed in unaffected family members. Enumeration of circulating white blood cells and platelets and examination of C-reactive protein showed no significant deviation from the normal range for Patient 2 with the RNASEH2A mutation and Patient 3 with the SAMHD1 mutation, even when Patient 2 had magnetic resonance imaging abnormalities and ongoing febrile episodes. Erythrocyte sedimentation rates fluctuated within the normal range for Patient 2, with some elevation, yet, were in the normal range during the second febrile episode when there were accompanying neurological abnormalities. These preliminary data suggest that ISG expression may be a more specific indicator of disease activity in comparison to standard inflammatory markers.
Introduction
A
AGS patients experience central nervous system (CNS) and systemic manifestations, including chronic cerebrospinal fluid pleocytosis and encephalopathy and may have intracranial calcifications, white matter lesions, and brain atrophy (Aicardi and Goutières 1984; Crow and Manel 2015). Associated systemic features include chilblains and idiopathic thrombocytopenic purpura (Rice and others 2007a; Crow and Manel 2015). In addition, disease activity can result in motor and cognitive delays, and ongoing systemic disease is associated with an estimated 8%–34% mortality rate (Rice and others 2007b; Crow and Manel 2015).
IFN signaling through the IFN-α/IFN-β receptor induces the expression of IFN-stimulated genes (ISGs), which encode proinflammatory and immunomodulatory proteins (Ivashkiv and Donlin 2014). Case–control studies have shown that AGS patients with confirmed mutations in any 1 of AGS1–AGS6 express higher levels of ISG15, RSAD2, IFI27, SIGLEC1, and IFI44L in comparison to healthy controls (Rice and others 2013). In light of increased ISG expression, a number of AGS patients develop systemic lupus erythematosus—an autoimmune disease with a type I IFN signature—in early childhood (Rice and others 2007a; Crow and Manel 2015).
Using quantitative real-time polymerase chain reaction (qPCR), we monitored the expression of select ISGs associated with AGS1–AGS6, namely ISG15, RSAD2, and IFI27, in the peripheral blood of 2 RNASEH2A AGS patients at 4 time points over a period of 18 months, and for 1 SAMHD1 AGS patient at 1 time point. We also monitored unaffected family members to characterize and compare the effects of carrying an AGS mutation on ISG expression. We provide evidence of anticipated elevated ISG expression for the AGS patients. We also compared peripheral blood ISG expression to a number of standard inflammatory markers: C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and platelet and white blood cell (WBC) counts to evaluate the potential for ISG expression to be used as a biomarker for AGS.
Materials and Methods
Subjects and ethics statement
We report on 3 AGS patients. Two siblings, Patient 1 and Patient 2, who have mutations in RNASEH2A. The unaffected mother and brother of Patients 1 and 2 with the RNASEH2A mutations are carriers for a c.158C>G (p.Pro53Arg) mutation in the RNASEH2A gene (AGS4), while the unaffected father is a carrier for a c.872G>A (p.Arg291His) mutation in the same gene. Both Patients 1 and 2 are compound heterozygotes for the mutations. Patient 3 is homozygous for a c.1411-2A>G (r.spl?) * mutation in the SAMHD1 gene (AGS5). Genetic analysis of the unaffected parents revealed heterozygosity for the SAMHD1 mutation in both parents.
Blood samples were obtained between February 2012 and September 2015. In addition, samples were also collected from the patients' unaffected family members. This study was approved by the Research Ethics Boards of The Hospital for Sick Children and the University Health Network (Toronto, ON, Canada). Signed consent was obtained from all participants.
Cells and reagents
Human lung adenocarcinoma epithelial A549 cells (ATCC, Manassas, VA) were maintained in RPMI-1640 with sodium bicarbonate and
RNA extraction and cDNA synthesis
Three milliliters of blood was drawn from patients and their unaffected family members following standard clinical methods into Tempus Blood RNA Tubes (Applied Biosystems, Waltham, MA) and processed using the Tempus Spin RNA Isolation Reagent Kit (Applied Biosystems) following the manufacturer's protocol. For tofacitinib-treated or tofacitinib-untreated samples, RNA from leukocytes was extracted using the RNeasy Mini Kit (Qiagen, Netherlands) according to the manufacturer's protocol. 1.5 × 107 A549 cells were treated overnight with 1 × 103 U/mL IFN-β-1a and RNA was also extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer's protocol. cDNA was generated using 1 μg of patient RNA or A549 cell RNA, random primers, and M-MLV reverse transcriptase (Invitrogen) according to the manufacturer's protocol.
Quantitative real-time polymerase chain reaction
qPCR was performed using LightCycler FastStart DNA Master SYBR Green PLUS I (Roche, Germany) and a LightCycler (Roche) according to the manufacturer's protocol and as previously described (Rogers and others 2012). Primers for HPRT1 [forward (5′ TCCTCCTCTGCTCCGCCACC 3′), reverse (5′ TCACTAATCACGACGCCAGGGCT 3′)], ISG15 [forward (5′ GCGGCTGAGAGGCAGCGAAC 3′), reverse (5′ TGCCCGCCAGCATCTTCACC 3′)], RSAD2 [forward (5′ GCCAAAACATCCTTTGTGCT 3′), reverse (5′ TGGCTCTCCACCTGAAAAGT 3′)], and IFI27 [forward (5′ ATCAGCAGTGACCAGTGTGG 3′), reverse (5′ GCCACAACTCCTCCAATCAC 3′)] were synthesized by ACGT Corporation (Toronto, ON, Canada). Standard curves for each gene were generated using cDNA from IFN-β-1a treated A549 cells. Data were analyzed using LightCycler Data Analysis Software (Roche).
Results
AGS patient histories
The 2 siblings, Patients 1 and 2, both aged 18 months at symptom onset, presented with a 4- to 6-week history of hemiparesis following a febrile episode associated with rash. In both siblings, initial magnetic resonance imaging (MRI) showed multifocal confluent white matter abnormalities (Fig. 1). Patient 1, a male, did not experience further neurological events. His younger sister, Patient 2, had a second episode at 4 months postonset, characterized by sialorrhea, decreased axial tone, recurrent fevers, and had progression of the white matter lesions on her MRI.
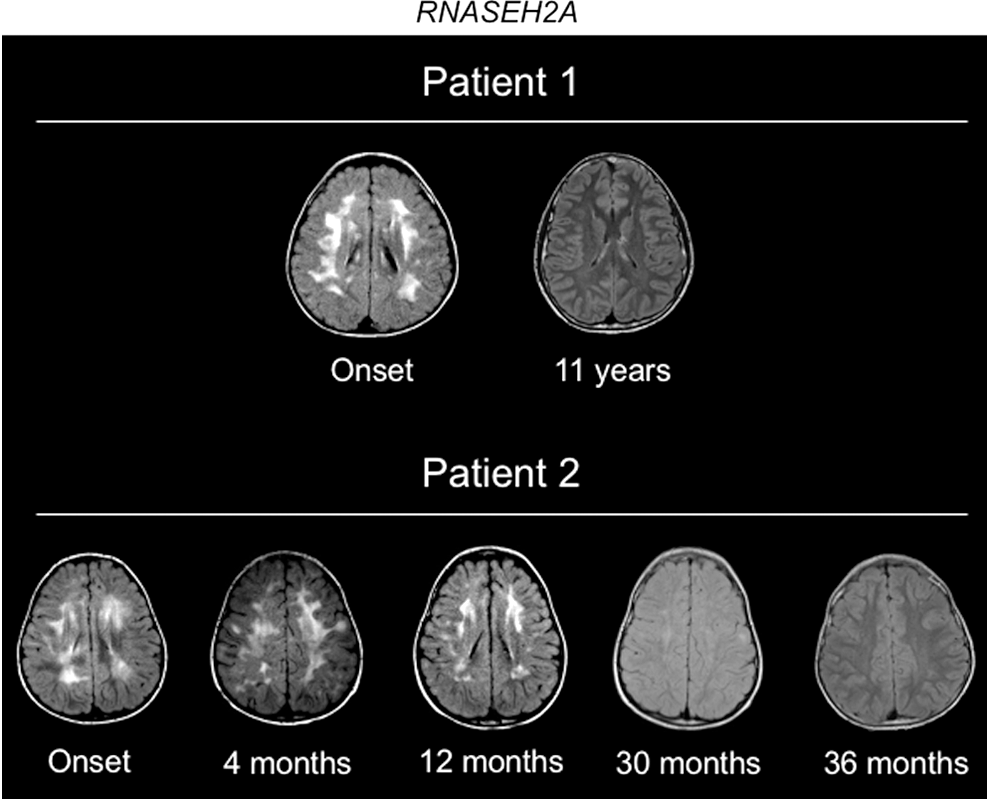
Brain MRIs for Patient 1 and Patient 2. Serial Axial T2 FLAIR images spanning onset and resolution of abnormalities. MRI, magnetic resonance imaging.
In both siblings MRI abnormalities regressed over time, yet, for Patient 2 worsened significantly at the time of an acute exacerbation and continued to show extensive T2 bright signal for several years after the initial acute event. Regression of the lesions occurred in conjunction with improvement of neurologic status (Fig. 1), but both have significant residual motor deficits, with severe cognitive impairment also present in Patient 2. For Patient 2, we conducted repeated rheumatological and inflammatory testing that revealed a modest elevation of WBCs and fluctuations in ESR, generally within normal range (Fig. 2A). Notably, at the time of the second febrile episode, ESR was within normal range. CRP levels and platelet numbers were consistently within normal range for Patient 2.
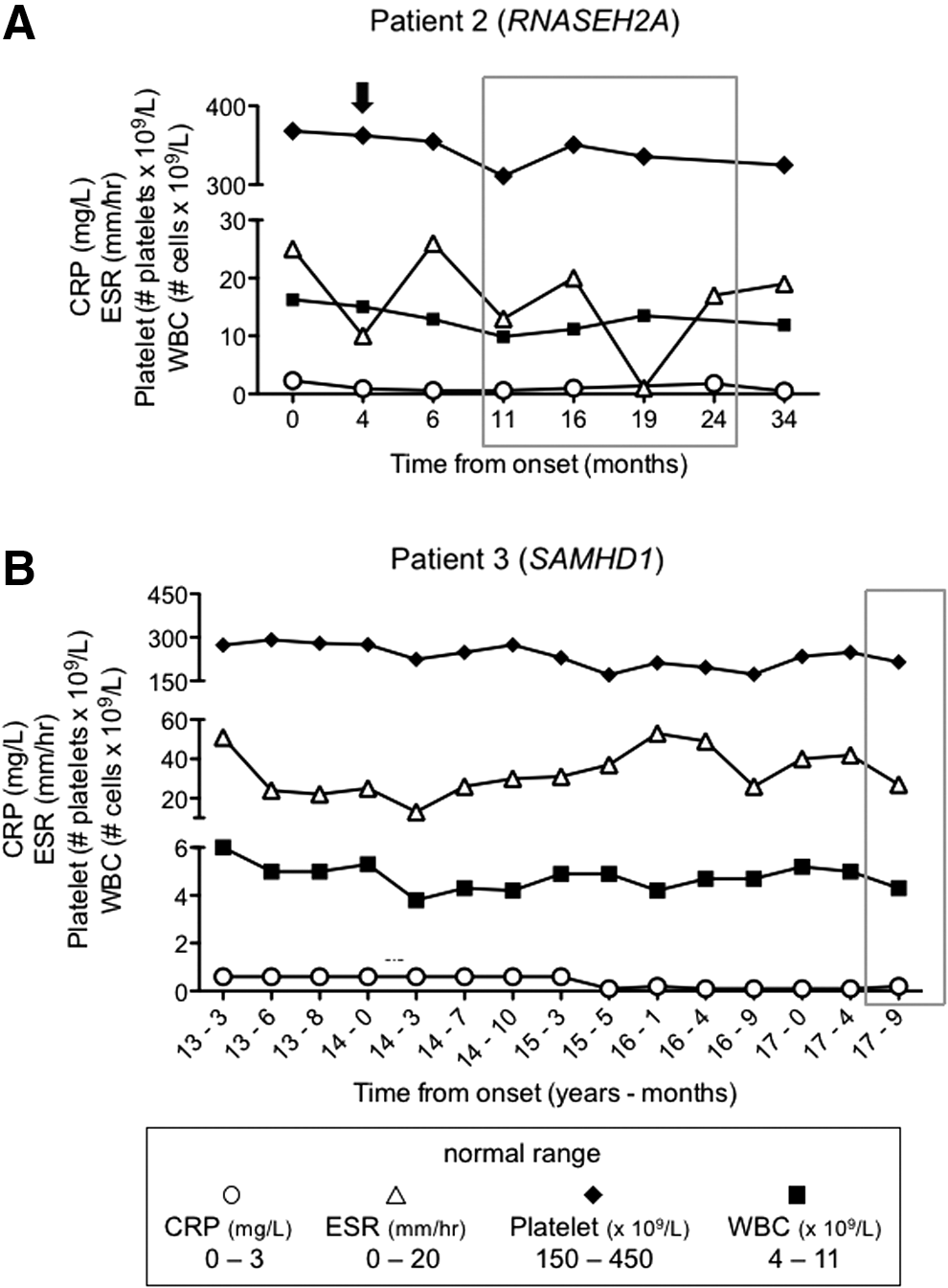
Standard blood markers for inflammation do not reflect persistent disease in AGS patients. Peripheral blood WBC (■) and platelet counts (♦), ESR (△), and CRP (◯) measurements were recorded over time for
Patient 3, presented at 1 year of age with chilblains, subsequently developed oral ulcers and arthritis at 9 years of age. Examination showed microcephaly, small stature, and developmental delay. MRI scans, performed at ages 12 years 9 months and 13 years 8 months, were unremarkable (data not shown). His polyarthritis was treated with etanercept, methotrexate, and intra-articular corticosteroids with excellent response, but severe chilblains persisted and led to tissue loss over the pinnae. His blood testing on therapy did show evidence for elevated ESR, but not CRP, with no significant effects on WBC or platelet counts while on immunosuppressive therapy (Fig. 2B).
AGS patients exhibit elevated peripheral blood ISG expression
Blood samples were collected from Patient 2 at 11, 16, 19, and 24 months postonset of AGS symptoms (Fig. 3A). At those times, we also obtained blood samples from Patient 1 (10 years 5 months–11 years 6 months postonset) and the siblings' unaffected mother and brother. By 10 years 5 months the gross MRI abnormalities in Patient 1 had regressed, with some residual abnormalities. MRI abnormalities in Patient 2 worsened and subsequently showed slow resolution over years (Fig. 1). When tested 11 months after the first onset of symptoms, Patient 2 exhibited ∼3.3-, 5.9-, and 7.3-fold higher levels of ISG15, RSAD2, and IFI27 expression, respectively, in her peripheral blood in comparison to her unaffected mother and brother, despite symptom resolution and no elevation in ESR, CRP, WBC, or platelets.
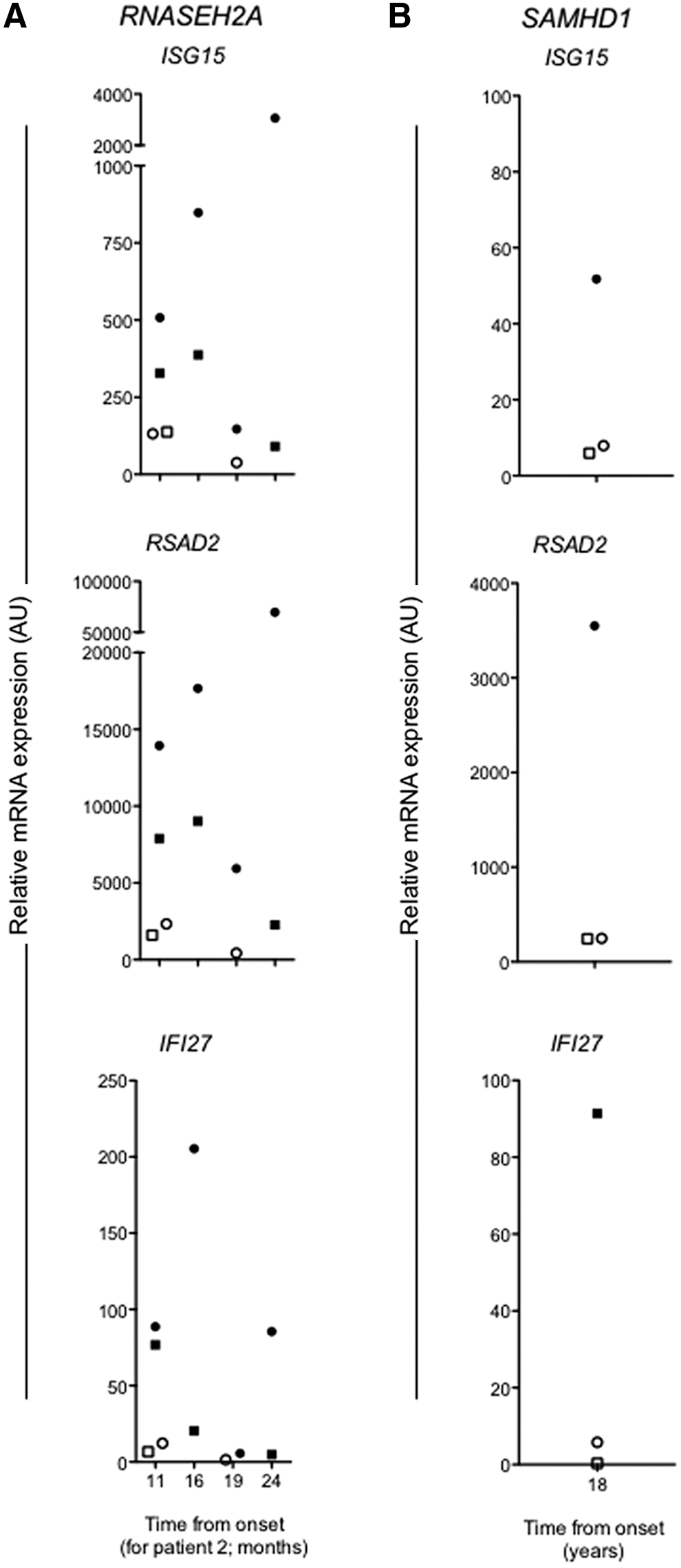
Elevated peripheral blood ISG expression in AGS patients.
Longitudinal evaluation of ISG expression in samples showed ongoing evidence for higher ISG expression in Patient 2, compared with Patient 1 and their unaffected mother. During this time period (11–24 months postsymptom onset), Patient 2 experienced occasional febrile episodes, but did not experience any new neurological events or new MRI lesions (Fig. 1). She continued to have severe neurological impairment. Interestingly, ISG expression levels decreased at 19 months postsymptom onset in Patient 2, the most significant decrease for IFI27 expression (36-fold), although still 3.3-fold greater than in the peripheral blood of the unaffected mother.
For Patient 3, we collected a blood sample at 18 years postonset of symptoms. At this time he continued to demonstrate chilblain lesions on his extremities and complained of 15–30 min of early morning stiffness and had flexion contractures of both fourth fingers, but no active arthritis. In comparison to his unaffected parents, Patient 3 had 6.5-fold greater ISG15 expression and more than 10-fold greater RSAD2 and IFI27 expression (Fig. 3B). As mentioned, only elevated ESR was observed, despite being on immunosuppressive therapy (Fig. 2B).
Discussion
Rice and others showed in an earlier case–control study that AGS caused by autosomal recessive mutations in RNASEH2A and SAMHD1 is associated with an ISG signature in the peripheral blood (Rice and others 2013). Prospective blood samples were collected from 4 to 16 patients with confirmed mutations in RNASEH2A and SAMHD1, respectively, and elevated ISG15, RSAD2, and IFI27 expression was observed compared with healthy control samples acquired from unrelated subjects (Rice and others 2013). In agreement, more recently Crow and others (2015) provided evidence for an almost 100% correlation between an elevated ISG expression signature and mutations in TREX1, RNASEH2A, RNASEH2C, SAMHD1, ADAR, and IFIH1 in a much larger cohort of 374 patients.
Both RNASEH2A and SAMHD1 encode proteins involved in nucleic acid metabolism. Specifically, RNASEH2A encodes the ribonuclease H2 subunit A, which is a component of the heterotrimeric type II ribonuclease H enzyme (RNAseH2) involved in the cleavage of ribonucleotides (Crow and others 2006b). SAMHD1 encodes sterile alpha motif (SAM) and histidine–aspartic (HD) domain containing deoxynucleoside triphosphate triphosphohydrolase 1, which has both phosphohydrolase and nuclease activities (Rice and others 2009; Goldstone and others 2011; Beloglazova and others 2013). Fibroblasts isolated from patients with confirmed mutations in RNASEH2A and SAMHD1 have an overabundance of RNA:DNA hybrids, which may contribute to the development of autoimmune responses and the elevated antiviral ISG signature associated with AGS (Lim and others 2015).
In contrast to previous studies, we tracked the ISG signature in the peripheral blood of 2 siblings with AGS (Patient 1 and 2) with compound heterozygous mutations in the RNASEH2A gene over a period of 13 months. We also collected blood samples from their unaffected mother and brother, who are carriers for a single mutation in RNASEH2A. In addition, we collected blood from 1 AGS patient (Patient 3) with a mutation in the SAMHD1 gene and both of his unaffected parents, who were also carriers for mutations in SAMHD1.
Consistent with previous reports (Rice and others 2007b), all 3 AGS patients in this study experienced severe manifestations of AGS-associated symptoms in early childhood—between 1 and 2 years of age—followed by varying degrees of symptom resolution. While the older sibling, Patient 1, experienced only a single episode of neuroinflammation, Patient 2 had a second, severe episode of encephalopathy accompanied by new MRI lesions months after the initial event, and was more severely clinically affected than her brother—she experienced severe global encephalopathy, ongoing central hypotonia, and marked motor weakness.
Although MRIs improved over time for Patients 1 and 2, and the severity of systemic disease diminished for Patient 3, we found persistently elevated ISG15, RSAD2, and IFI27 expression in their peripheral blood relative to their unaffected family members. Notably, ISG expression was higher in Patient 2 than in Patient 1, which may correlate with differing stages of CNS disease and age (Rice and others 2013).
Furthermore, we were able to compare our peripheral blood ISG data with standard serum testing for inflammation, including CRP, ESR, and platelet and WBC counts. We found that ESR and WBC counts were mildly elevated in Patient 2; however, only ESR readings correlated with changes in peripheral blood ISG expression. Also in Patient 2, ESR was within the normal range at 4 months postonset when the girl experienced a second episode of CNS symptoms.
Altogether, our data further support the evidence of an ISG signature in the peripheral blood of AGS patients and provide preliminary insights into changes in ISG expression in relation to different stages of CNS disease progression and resolution. Moreover, our data suggest that ISG expression may be a more specific marker for disease than ESR and CRP alone and could be used to complement current readouts for inflammation in patients with IFN-mediated diseases. ISG expression as an early childhood screen for suspected AGS may allow for rapid identification of this disorder and presents a potential target for future drug therapies that target an IFN response. We suggest that scrutiny of the IFN-sensitive gene transcriptome in the context of episodes of disease activity is worthy of further evaluation.
Footnotes
Acknowledgment
E.N.F. is a tier 1 Canada Research Chair.
Author Disclosure Statement
No competing financial interests exist.