
Abstract
Transfusion of packed red blood cells (PRBCs) modulates patients' immune responses and clinical outcomes; however, the underpinning mechanism(s) remain unknown. The potential for PRBC to modulate myeloid dendritic cells (mDC) and blood DC antigen 3 was assessed using an in vitro transfusion model. In parallel, to model processes activated by viral or bacterial infection, toll-like receptor agonists polyinosinic:polycytidylic acid or lipopolysaccharide were added. Exposure to PRBC upregulated expression of CD83 and downregulated CD40 and CD80 on both DC subsets, and it suppressed production of interleukin (IL)-6, IL-8, IL-12, tumor necrosis factor-α, and interferon-gamma-inducible protein-10 by these cells. Similar effects were observed when modeling processes activated by concurrent infection. Furthermore, exposure to PRBC at date of expiry was associated with more pronounced effects in all assays. Our study suggests PRBC have an impact on recipient DC function, which may result in failure to establish an appropriate immune response, particularly in patients with underlying infection.
Introduction
T
PRBC is one of the major blood components derived from donor whole blood, where the RBCs are separated from platelets and plasma, and are depleted of leukocytes via filtration (Council of Europe 2008). PRBC are routinely stored for up to 42 days (date of expiry) in the presence of storage additive solution (saline-adenine-glucose-mannitol) before transfusion. During ex vivo storage at 4°C (Council of Europe 2008), RBC within the product undergo significant modifications that can alter cellular function and viability (Mitrofan-Oprea and others 2007; Glynn and others 2016). This phenomenon was originally described by Gabrio and others (1954) who demonstrated that refrigerated RBC age more rapidly than those in vivo. While storage-related morphological and biochemical modifications in stored PRBC are well documented (Van de Watering 2011), whether these changes contribute to worse patient outcomes remains controversial, with correlation of stored PRBC with poor patient outcomes in some studies (Mynster and Nielsen 2001; Leal-Noval and others 2003; Dunn and others 2012; Dhabangi and others 2015; Lee and others 2015), but not others (Yap and others 2008; Kaukonen and others 2014; Lacroix and others 2015; Steiner and others 2015; Yamal and others 2015; Heddle and others 2016). Recent prospective randomized controlled trials did not find an association between storage duration of PRBC and adverse clinical outcomes (Kaukonen and others 2014; Lacroix and others 2015; Steiner and others 2015; Yamal and others 2015; Heddle and others 2016). However, conclusions that can be drawn regarding transfusion of PRBC near date of expiry are limited as these randomized trials are restricted due to ethical considerations. Due to the study design, the average storage duration of PRBC that were considered “old” in these studies only approximates half of the actual storage period. The impact of transfusing stored PRBC near date of expiry on modulating patient outcomes remains a highly controversial issue in transfusion medicine.
As cellular processes associated with transfusion of stored PRBC remains undefined, in vitro and in vivo models have been used to study and understand the potential for PRBC to modulate recipients' immune response and impact patient outcomes. In vitro studies demonstrate modulation of monocyte (Muszynski and others 2011) and overall leukocyte (Biedler and others 2002; Karam and others 2009; Schneider and others 2009) inflammatory responses following exposure to PRBC or PRBC components. These studies consistently report suppression of tumor necrosis factor (TNF)-α and augmentation of interleukin (IL)-10 responses. Increased monocyte erythrophagocytosis of stored PRBC has also been reported (Veale and others 2014). Transfusion-related immune modulation is not limited to innate cells, with exposure to PRBC reported to increase induction of CD25+ regulatory T lymphocytes, reduce the number of responder T lymphocytes (Baumgartner and others 2009), and suppress B and T lymphocyte proliferation (Bernard and others 2010; Long and others 2013). To date, studies assessing the role of dendritic cells (DC) in transfusion outcomes are limited. A murine model using non-leukodepleted PRBC reported CD200+ DC may play a role in modulating transfusion-associated tumor growth (Clark and others 2008). Erythrophagocytosis by murine plasmacytoid DC has been reported, particularly in combination with polyinosinic:polycytidylic acid (polyI:C)-driven inflammation (Richards and others 2016). Murine studies have reported that splenic DC are important for outcomes in terms of alloimmunization (Hendrickson and others 2007; Calabro and others 2016). Despite these murine studies, our understanding of whether DC play a role in modulating patient outcomes following PRBC transfusion remains largely undefined.
DC play a crucial and central role in the immune system at the interface of innate and adaptive immune responses (Hoebe and others 2004). DC are classified into 2 major lineages, myeloid DC (mDC) and plasmacytoid DC (Wu and Liu 2007), which are further divided into myeloid blood DC antigen (BDCA)1+ (CD1c+), myeloid BDCA3+ (CD141+), plasmacytoid BDCA2+ (CD303+), and plasmacytoid BDCA4+ (CD304+) DC (Dzionek and others 2000; MacDonald and others 2002). These cells have specialized functions shaped by the differential expression of numerous pattern recognition receptors (PRRs) including toll-like receptors (TLRs) and C-type lectin receptors. These receptors have the capacity to recognize pathogen-associated molecular patterns, or damage-associated molecular patterns (DAMPs), which enable DC to detect invading pathogens, or damaged cell components respectively. mDC are the more abundant lineage and they predominantly express PRR primarily for recognition of bacteria and subsequent antigen presentation via major histocompatibility complex (MHC) class II. Interestingly, a rare subset of mDC, BDCA3+ DC are equipped with PRR skewed toward recognition of viruses, and these cells are specialized in dead cell-associated antigen cross-presentation (via MHC class I) (Jongbloed and others 2010; Cohn and others 2013). Following pattern recognition, DC become activated and undergo phenotypic changes essential for antigen presentation including expression of co-stimulatory molecules and secretion of cytokines and chemokines (Banchereau and Steinman 1998; Yao and others 2002). Perturbation of these DC processes has the potential to significantly affect host immune responses (Elia and others 2008; Mancino and others 2008), which may contribute to poor outcomes in transfusion recipients.
While PRBC have been reported to modulate cells of the innate and adaptive immune response, the impact of transfusion of DC responses remains largely unexplored. As central cells in the immune response, we investigated whether mDC and the specialized subset BDCA3+ DC were modulated following exposure to PRBC using an in vitro transfusion model. Further, the impact of PRBC on mDC and BDCA3+ DC capacity to respond adequately to concurrent immune challenges such as infection was modeled using TLR agonists polyI:C (“viral”) or lipopolysaccharide (LPS; “bacterial”). Herein, we sought to address the hypothesize that DC are particularly vulnerable to modulation by transfusion due to their multifaceted role in pattern recognition, phagocytosis, activation, and signaling the adaptive immune response, and propose changes in DC phenotype and function are 1 mechanism underpinning transfusion-related immune modulation.
Materials and Methods
Ethics
The Australian Red Cross Blood Service (Blood Service) Human Research Ethics Committee and University of Queensland School of Medicine Low Risk Ethical Review Committee approved this study.
Blood components
Leukodepleted PRBC units were obtained from the Blood Service. Whole blood units (450 ± 45 mL) were collected into top-and-bottom bags containing citrate phosphate dextrose (66.5 mL; Macopharma, Mouvaux, Nord, France), and processed within 24 h of collection according to standard Blood Service protocols based on the Council of Europe “Guidelines for the preparation, use and quality assurance of blood components” (Council of Europe 2008). Briefly, whole blood units were spun (3,640 g, 10 min, 22°C) and RBC were separated into the bottom bag containing storage additive (saline, adenine, glucose and mannitol solution; 105 mL) using a MacoPress component extractor (Macopharma). PRBC units (≥185 mL; 1.0 × 106 leukocytes per unit) were stored at 2°C–6°C, and used “fresh” (D2) or “stored” (D42, date of expiry).
In vitro whole blood model of transfusion
An in vitro whole blood model was used to assess potential immune modulation (mDC and BDCA3+ DC surface antigen expression and inflammatory mediator production) in a transfusion recipient. This assay has been established to more closely model transfusion by using a whole blood approach that facilitates cell-to-cell interactions of the PRBC with leukocytes subsets without the need for cell isolation procedures involving density gradients or cell separation (Perros and others 2015). Fresh whole blood samples from healthy volunteers were collected in ethylenediaminetetraacetic acid (EDTA) blood collection tubes [Becton Dickinson (BD) Biosciences, NJ] and served as “transfusion recipient” in our model. EDTA was selected as the preferred anticoagulant in this model as it has shown to be consistent and suitable for analysis of cytokine inflammatory responses (Thavasu and others 1992; Flower and others 2000). A 25% blood replacement volume transfusion (representing 2–3 U) was modeled through culturing “recipient” whole blood (6 mL) with ABO compatible PRBC [D2 (fresh) or D42 (stored) PRBC (from different units); 3 mL] and Roswell Park Memorial Institute (RPMI) 1640 media (3 mL; Invitrogen, CA) for 5.5 h (37°C, 5% CO2). PolyI:C (50 μg/mL, high molecular weight; Invitrogen) or LPS (1 μg/mL; Sigma-Aldrich, St. Louis, Missouri) was added in duplicate tubes to model the processes activated by concurrent viral (polyI:C) or bacterial (LPS) infections. Matched “no transfusion” controls were included in parallel with PRBC volume replaced with RPMI 1640 (included polyI:C and LPS only controls). For experiments assessing intracellular mediators, GolgiPlug (1 μg/mL; BD Biosciences) was added for the last 4.5 h. Cells were harvested and supernatant collected from cultures without GolgiPlug [stored at −80°C for later analysis using cytometric bead array (CBA)]. RBC were lysed [10 min room temperature (RT), FACS lyse; BD Biosciences] and residual leukocytes washed thrice in 3% fetal bovine serum in phosphate-buffered saline (3% FBS/PBS, both from Invitrogen) before staining for flow cytometry.
Assessment of DC surface antigen expression and intracellular inflammatory mediator production
Leukocytes were stained (15 min, 4°C) with the following DC panel of mouse anti-human mAb (all from BD Biosciences unless stated otherwise): “lineage negative”-fluorescein isothiocyanate (Lin−-FITC; CD3, CD14, CD19, CD20, CD34, CD56), CD45− peridinin chlorophyll (PerCP), CD11c-brilliant violet (BV)421, and BDCA3− allophycocyanin (APC; Miltenyi Biotec, Bergisch Gladbach, Germany). For assessment of surface antigen expression, leukocytes were also stained with CD40-phycoerythrin (PE) and CD80-BV510 or CD83-PE and CD86-BV510. Cells were washed twice (3% FBS/PBS) and resuspended in 3% FBS/PBS for flow cytometric analysis. For assessment of intracellular mediators, following staining with the DC panel outlined above, leukocytes were permeabilized (10 min, RT; FACS Perm; BD Biosciences) and labeled with intracellular PE-conjugated mAb targeting IL-6, IL-8, IL-12, TNF-α and, IL-10 (LPS panel) or, interferon-gamma-inducible protein (IP)-10 (polyI:C panel) for 30 min (RT, dark). Cells were washed (3% FBS/PBS) and resuspended in cell stabilizing fixative (BD Biosciences) for flow cytometry.
Flow cytometry gating strategies
mDC were gated as Lin−CD45+CD11c+ and BDCA3+ DC as Lin−CD45+CD11c+BDCA3+. Briefly, adopting gating strategy described in our previous study (Ki and others 2017): Side scatter versus CD45 was used to exclude granulocytes and lymphocytes, and selected mononuclear cells were displayed as Lin versus CD11c with Lin−CD11c+ cells selected as mDC. The selected mDC population was then further selected for gating of the BDCA3+ DC subset. Median fluorescent intensity (MFI) for each surface antigen and inflammatory mediator expressed by mDC and BDCA3+ DC was determined and all statistical analyses were performed on raw data (see Statistical Analysis section below).
Data presented in figures were derived from a 2-step approach. First, PRBC “transfusion” data were normalized (using a ratio of the MFI) to the matched “no transfusion” control. Specifically, data derived from “recipient” whole blood+D2 or D42 PRBC was normalized to the matched “recipient” whole blood only (ie, “no transfusion”) control. Similarly for the whole blood approach modeling processes activated by infection, data from “recipient” whole blood+D2 or D42 PRBC+polyI:C or LPS were normalized to the matched “recipient” whole blood+polyI:C or LPS only control. The MFI ratios were then calculated as binary logarithm (log2). This approach effectively “equalizes” the data around zero to display a fold change. A 2-fold increase is therefore +1 and a 2-fold decrease is therefore as −1 compared to the matched control. This approach establishes a baseline for each donor to account for donor-to-donor variation (representative data Supplementary Figs. S1 and S2; Supplementary Data are available online at
Assessment of overall leukocyte inflammatory response
Concentrations of inflammatory mediators [IL-1α, IL-1β, IL-8, IL-6, IL-10, TNF-α, IL-12p70, IP-10, interferon (IFN)-α, IFN-γ], monocyte chemoattractant protein (MCP)-1, macrophage inflammatory proteins (MIP)-1α, MIP-1β were assessed in culture supernatant from the transfusion model (n = 16 PRBC only, n = 8 PRBC+polyI:C, n = 8 PRBC+LPS) using CBA according to manufacturer's directions (BD Biosciences). Concentrations were determined from standard curves that ran in parallel.
Assessment of erythrophagocytosis
D2 and D42 PRBC were labeled with FITC-conjugated-function-spacer-lipid (50 μg/mL, FSL-FLRO4; Kode Biotech Materials Limited, Auckland, New Zealand) as described (Blake and others 2011; Ki and others 2016). Briefly, an equal volume of PRBC and FSL-FLRO4 were vortexed and incubated (37°C, 1 h, dark), then washed and resuspended in PBS (500 μL). Peripheral blood mononuclear cells (PBMCs), isolated using Ficoll-Paque (GE Healthcare, Munich, Bavaria, Germany) density gradient centrifugation were stained with HLA-DR-PerCP, BDCA3-APC, CD11C-BV421, and CD14-V500 (15 min, 4°C, dark). FSL-FLRO4-PRBC (D2 or D42) were mixed with PBMC (10:1 ratio), and incubated (30 min, 37°C, 5% CO2) before noninternalized PRBC were lysed (FACS lyse; BD Biosciences). Phagocytosis of PRBC (expressed as percent positive and MFI) was determined by flow cytometry in gated monocyte (CD14+), mDC (CD14−DR+CD11c+), and BDCA3+ DC (CD14− DR+CD11c+BDCA3+) populations.
Flow cytometry
Three laser FACSCanto™ II flow cytometer (BD Biosciences) was used and data analyzed using FCS express V5 (De Novo, Los Angeles) or FCAP Array software (BD Biosciences).
Statistical analysis
GraphPad Prism (GraphPad Software, Santiago) was used for all statistical analyses and representation of graphs. Analysis of variance (ANOVA) with Tukey's post-tests were used on raw data to assess the differences in surface antigen expression and inflammatory mediator production following PRBC exposure. A separate ANOVA was performed for each experimental outcome, (1) assessment of changes in DC responses to PRBC transfusion [no transfusion (ie, “recipient” whole blood only) versus D2 PRBC versus D42 PRBC], (2) assessment of changes in DC responses to poly I:C in the presence of PRBC [no transfusion (ie “recipient” whole blood only)+polyI:C versus D2 PRBC+polyI:C versus D42 PRBC+polyI:C], and (3) assessment of changes in DC responses to LPS in the presence of PRBC [no transfusion (ie “recipient” whole blood only)+LPS versus D2 PRBC+LPS versus D42 PRBC+LPS]. Phagocytosis data were compared using paired t-tests. For all analyes P < 0.05 was considered significant.
Results
Exposure to PRBC modulated mDC and BDCA3+ DC surface antigen expression
An in vitro whole blood model of transfusion was used to assess the impact of PRBC on activation and maturation of mDC and BDCA3+ DC. Changes in cell-specific responses of these DC subsets were investigated using multicolor flow cytometry. Exposure to PRBC alone downregulated mDC expression of co-stimulatory molecules CD40 (P < 0.0001) and CD80 (P = 0.019), and upregulated expression of CD83 (P < 0.0001; Fig. 1A–C). When polyI:C was cocultured with PRBC to model the processes activated by viral infection, mDC expression of CD80 (P = 0.006) was downregulated and CD83 was upregulated (P = 0.006, Fig. 1C) and there was no change in expression of CD40 or CD86 compared to polyI:C alone (Fig. 1A, D). In the model of bacterial infection, mDC expression of CD86 (P = 0.030, Fig. 1D) was downregulated, and there was no change in CD40, CD80, or CD83 (Fig. 1A–C).

Assessment of mDC and BDCA3+ DC expression of maturation and co-stimulatory molecules following exposure to D2 and D42 PRBC. Data represent flow cytometric analysis of mDC
For BDCA3+ DC, exposure to PRBC alone downregulated expression of CD80 (P = 0.006, Fig. 1F) and upregulated CD83 (P < 0.0001, Fig. 1G), with no changes in expression of CD40 or CD86 (Fig. 1E, H). In the presence of polyI:C, exposure to PRBC was associated with reduced BDCA3+ DC expression of CD86 (P = 0.006) (Fig. 1H). When LPS was used to model the processes activated by bacterial infection, BDCA3+ DC expression of CD86 (P = 0.0006) was suppressed and CD83 (P = 0.005) was increased in the presence of PRBC (Fig. 1G, F). For both DC subsets, modulation of maturation and co-stimulatory molecules were particularly evident following exposure to stored (D42) PRBC (Tukey's post-tests).
Exposure to PRBC downregulated mDC and BDCA3+ DC production of inflammatory mediators
Using the same whole blood approach we further assessed the impact of PRBC on cytokine and chemokine production by mDC and BDCA3+ DC using intracellular staining. Two panels of inflammatory mediators were used based on responses to viral or bacterial stimuli. Expression of inflammatory mediators was not significantly altered following exposure to PRBC alone for mDC (Fig. 2A–F). When PRBC were cocultured with polyI:C, mDC production of IL-6 (P = 0.004), IL-8 (P = 0.003), IL-12 (P = 0.005), and TNF-α (P = 0.003) was reduced (Fig. 2A–D). Similarly, coculture of PRBC with LPS resulted in suppression of proinflammatory cytokines IL-6 (P = 0.002) and TNF-α (P = 0.015) (Fig. 2A, D).
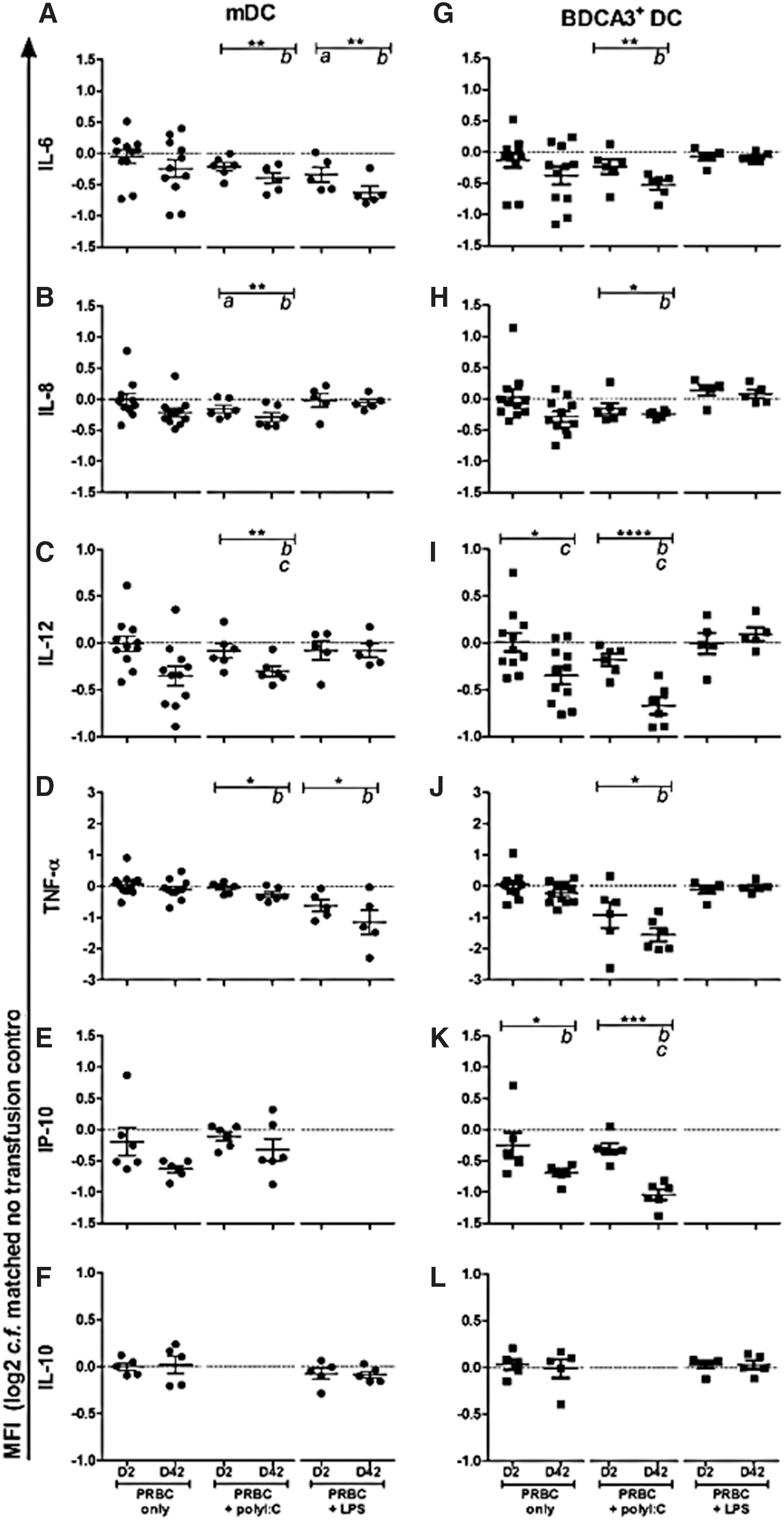
Assessment of mDC and BDCA3+ DC cytokine and chemokine production following exposure to D2 and D42 PRBC. Data represent flow cytometric analysis of mDC
For BDCA3+ DC, however, we found a different response profile. Expression of IL-12 (P = 0.028) and IP-10 (p = 0.047) were suppressed following exposure to PRBC alone (Fig. 2J, K). Coculture of PRBC with LPS did not result in altered production of inflammatory cytokines, however, IL-6 (P = 0.006), IL-8 (P = 0.015), IL-12 (P < 0.0001), TNF-α (P = 0.019), and IP-10 (P = 0.0002) production were significantly suppressed following coculture of PRBC and polyI:C (Fig. 2G–K). For both subsets, changes in production of inflammatory mediators were particularly evident following exposure to D42 PRBC (Tukey's post-tests).
Exposure to PRBC modulated the overall leukocyte inflammatory response
The use of the whole blood approach also allowed us to assess the impact of PRBC on the overall cytokine and chemokine production, representing changes from the mixed leukocyte population as a whole. Exposure to PRBC alone did not result in modulation of the overall leukocyte inflammatory response (Fig. 3A–F). In the presence of polyI:C, IL-8 production was augmented following exposure to D2 PRBC (P = 0.001, Fig. 3A). In the presence of LPS, exposure to PRBC upregulated IL-1β (P = 0.003), IL-6 (P = 0.001), IL-8 (P = 0.020), MIP-1α (P = 0.020), MIP-1β (P = 0.003), and downregulated IP-10 (P = 0.014) production (Fig. 3A–F). Changes in production of inflammatory mediators were particularly evident following exposure to D42 PRBC (Tukey's post-tests). No changes were observed in overall levels of IL-1α, IL-10, IL-12p70, TNF-α, MCP-1, IFN-α, or IFN-γ following exposure to PRBC alone, PRBC with polyI:C nor PRBC with LPS (data not shown).
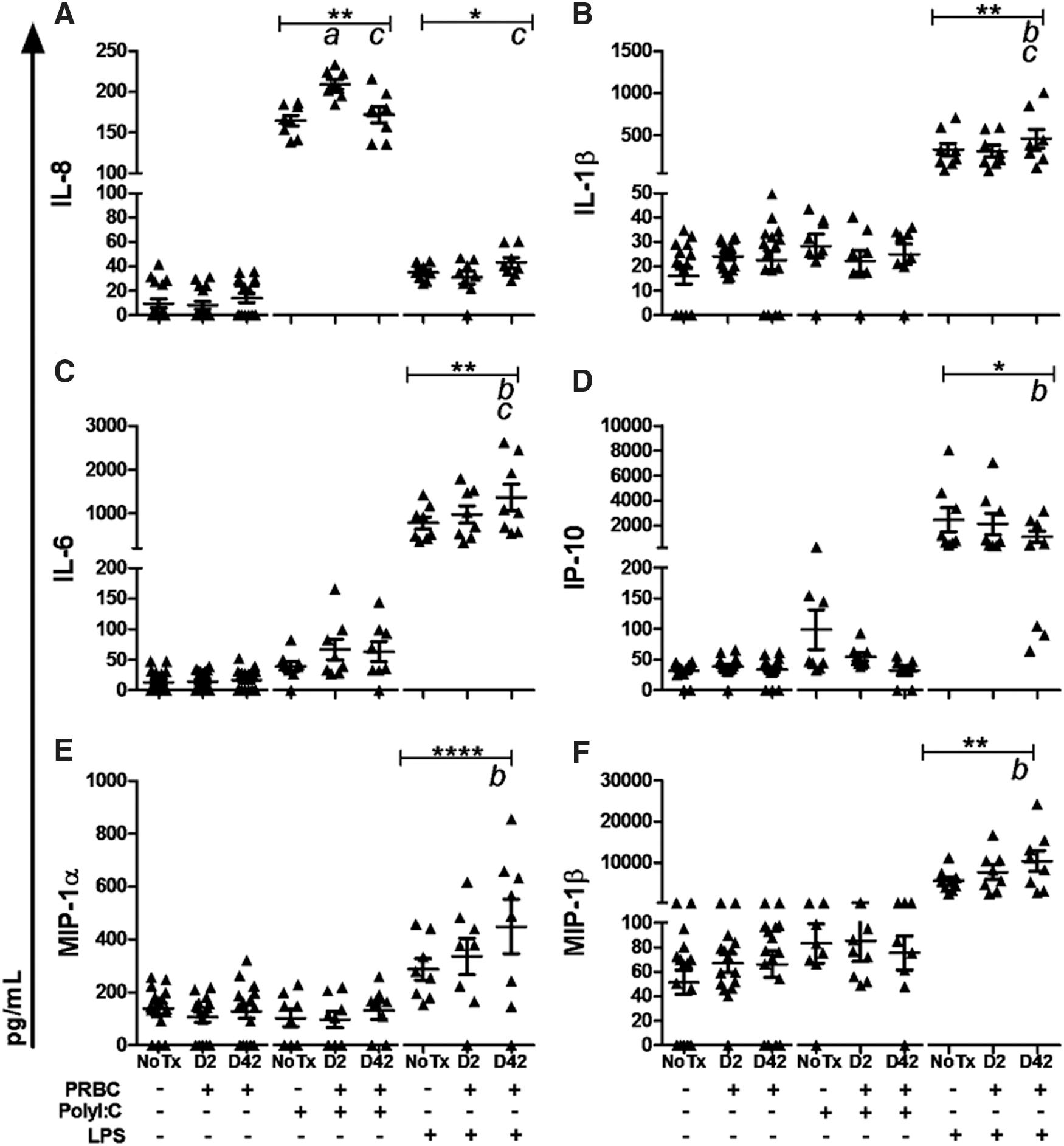
Assessment of the overall leukocyte inflammatory response following exposure to D2 and D42 PRBC.
PRBC storage was associated with increased erythrophagocytosis
We assessed whether phagocytosis of PRBC may be a potential mechanism driving changes in mDC and BDCA3+ phenotype in the transfusion model outlined above. While increased uptake of stored PRBC by monocytes has been reported, whether mDC, and BDCA3+ DC, contribute to PRBC clearance was unknown. We found that while monocytes were the primary cells associated with PRBC phagocytosis, mDC and BDCA3+ DC both phagocytosed D2 and D42 PRBC (Fig. 4A). For all 3 myeloid subsets assessed, D42 PRBC were phagocytosed more than D2 PRBC (Fig. 4B).
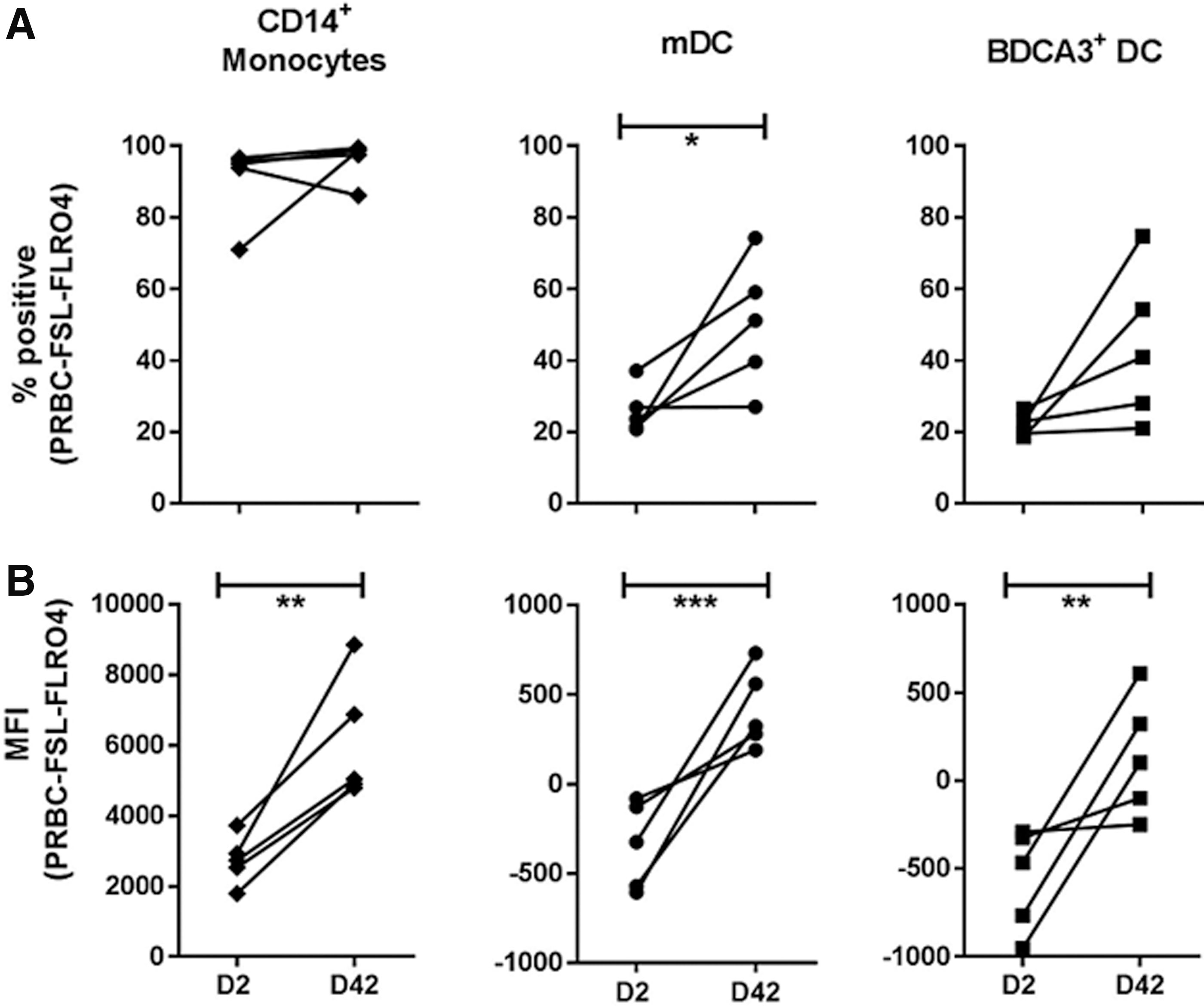
Assessment of erythrophagocytosis by mDC and BDCA3+ DC.
Discussion
Transfusion of PRBC has been reported to modulate recipients' immune responses. However, little is known about the mechanisms underpinning transfusion-related immune modulation. As DC play a central role in the initiation and regulation of both innate and adaptive immunity, we hypothesized that PRBC transfusion modulates key DC processes, impeding the capacity of these cells to adequately respond to subsequent immune challenges. We observed significant modulation of mDC and BDCA3+ DC phenotype following exposure to PRBC in a number of transfusion models (PRBC only and PRBC ± polyI:C/LPS). Of particular importance for outcomes in transfusion recipients, we found changes to DC phenotype were predominantly found in our models of infection, and stored PRBC had a more pronounced effect than fresh PRBC. This study provides evidence that while PRBC transfusion is lifesaving, the potential for patients with concurrent infection to develop worse outcomes following transfusion due to the impeded DC function needs to be considered.
DC maturation and co-stimulation are an essential process for downstream immune initiation and regulation (Banchereau and Steinman 1998). We first assessed the effect of PRBC on mDC and BDCA3+ DC maturation and co-stimulatory surface antigen profile. We report augmentation of maturation molecule CD83 and suppression of co-stimulatory molecules (CD40, CD80, and CD86) following exposure to PRBC. CD83 is upregulated following exposure to pathogens (Banchereau and others 2000) or TLR agonists including polyI:C (TLR3) and LPS (TLR4) (Hemont and others 2013). Our finding that exposure to PRBC alone upregulates CD83 suggests that either RBCs themselves, or substances within the PRBC product have the capacity to stimulate DC maturation. Although CD83 increased, the co-stimulatory molecules were predominantly downregulated following exposure to PRBC. Given the complexity of the PRBC product and the changes that occur during storage, it is likely that multiple DC receptors are engaged resulting in differential expression of maturation and co-stimulatory molecules.
In addition to expression of maturation and co-stimulatory molecules, a balanced release of cytokines and chemokines by DC is also crucial for directing multiple immune pathways (Calabro and others 2016). We found minimal modulation of cytokine responses following exposure to PRBC alone, however, both mDC and BDCA3+ DC responses to polyI:C and LPS were suppressed in the presence of PRBC. Together poor co-stimulation and lack of cytokine and chemokine production may be associated with inadequate T and B lymphocyte responses (Baumgartner and others 2009; Bernard and others 2010; Long and others 2013) and our data suggest PRBC transfusion has the potential to impair downstream lymphocyte responses.
We hypothesized that changes in mDC and BDCA3+ DC phenotype may be in part due to phagocytosis of PRBC. Indeed we report that both mDC and BDCA3+ DC play a role in RBC uptake, and stored PRBC were phagocytosed more than fresh PRBC. Therefore, in addition to uptake by monocytes (THP-1) as reported previously (Veale and others 2014), mDC and BDCA3+ DC both preferentially recognize and remove stored PRBC. This is likely attributed to the significant changes in PRBC that occur during routine storage that result in expression of a number of DAMP (Bosman and others 2008). It has been reported that inflammation enhances erythrophagocytosis (Hendrickson and others 2007; Richards and others 2016) and the modulation of mDC and BDCA3+ DC phenotype observed in this study, particularly in the models processes associated with infection, may be the result of increased erythrophagocytosis.
In addition to studying mDC and BDCA3+ DC specific responses, we investigated the overall response of the peripheral blood leukocytes in the culture. This largely represents the expected changes in the response of the more abundant leukocyte populations in patient's plasma following PRBC transfusion. We observed minimal impact on the overall leukocyte inflammatory response in the presence of PRBC alone, and PRBC cocultured with polyI:C. However, PRBC augmented production of IL-1β, IL-8, MIP-1α, and MIP-1β and, suppression of IP-10 following PRBC exposure when modeling processes associated with bacterial infection. We found it interesting that the overall leukocyte inflammatory response was largely augmented by exposure to PRBC whereas the specific cellular responses of mDC and BDCA3+ DC were attenuated. These results highlight the potential for mechanisms associated with specific cell subsets to be missed when only assessing “overall inflammation,” which largely represent the response of numerous cells subsets such as neutrophils, lymphocytes, and monocytes.
We studied not only on mDC but also the rarer subset of mDC, BDCA3+ DC, as they have a specialized and unique role in the immune response. While mDC express PRR predominantly associated with recognition of bacteria, the PRR profile of BDCA3+ DC is skewed toward recognition of viruses. Following receptor ligation, mDC present antigen via the standard MHC class II pathway, but BDCA3+ are more superior in cross-presentation of dead cell-associated antigens onto MHC class I. BDCA3+ DC have been shown to be important in CD8+ T lymphocyte responses (Jongbloed and others 2010; Cohn and others 2013) and have been identified as a key cell involved in anti-tumor immunity (Hildner and others 2008). Whether BDCA3+ DC play a role in cancer recurrence in transfusion recipients would be of interest for future studies.
In summary, we report that exposure to PRBC modulated the cellular responses of both mDC and the specialized subset BDCA3+ DC. Importantly, we found modulation of DC inflammatory response was particularly evident in models combining transfusion and infection, and, a more prominent affect was observed following exposure to stored PRBC. These results suggest that patients who receive stored PRBC, particularly those with underlying infectious complications, may fail to mount an appropriate immune response precipitating worse patient outcomes.
Footnotes
Acknowledgments
K.K.K. is supported by an Australian Post-graduate Award from the University of Queensland and top-up scholarship from the Alex Steele Young Memorial Lions Foundation. Australian governments fund the Australian Red Cross Blood Service for the provision of blood, blood products, and services to the Australian community. We would like to thank Professor Stephen Henry (Kode Biotech Materials Limited) for provision of FSL constructs for this study.
Author Disclosure Statement
No competing financial interests exist.