
Abstract
Obesity is a prominent risk factor for colorectal cancer (CRC). One mechanism by which obesity promotes the development of CRC is by generating a chronic, low-grade state of colonic inflammation. Interleukin-1β (IL-1β), a proinflammatory cytokine often elevated in obesity, is known to activate several procarcinogenic signaling pathways that are implicated in colonic carcinogenesis. We therefore sought to define the role of IL-1β in mediating some of the early biochemical and molecular events leading up to obesity-promoted CRC. Twenty-five wild-type (WT) C57BL/6J mice and 24 lacking a functional IL-1 receptor (IL1R−/−) were each randomized to either low-fat or high-fat diets, resulting in lean and obese mice. Compared to WT lean controls, WT obese mice displayed 30%–80% greater concentrations of IL-1β and tumor necrosis factor-α (TNF-α) in the colonic mucosa (IL-1β: P = 0.04; TNF-α: P < 0.05), activation of the Wnt signaling cascade [evidenced by a 2-fold increase in colonic crypt cells displaying intranuclear β-catenin (P < 0.03)], and a significant expansion of the proliferation zone of the colonic crypt (P < 0.04). These obesity-induced alterations in colonic cytokines, Wnt signaling, and proliferation were absent in the obese IL1R−/− mice. In the absence of IL-1 signaling, obesity-induced elevations of colonic IL-1β, TNF-α, Wnt activation, and enhanced epithelial proliferation no longer occur. These observations underscore the important mechanistic roles that IL-1 signaling appears to play in mediating the procancerous effects of obesity in the colon, thereby identifying a potential target for future strategies aimed at chemoprevention.
Introduction
E
It has been known for over 2 decades that obesity produces a chronic, low-grade inflammatory state that is evident in the blood and adipose tissue, but it is only recently that observations have demonstrated that this inflammatory milieu also resides in the colons of obese animals (Jain and Bird 2010; Flores and others 2012; Liu and others 2012). The evidence to date suggests that this obesity-induced inflammation in the colon plays an important [although not necessarily exclusive (Allott and Hursting 2015)] role in mediating the increased risk of colonic carcinogenesis.
Although a few studies have highlighted the role that tumor necrosis factor-α (TNF-α) may play in mediating the procancerous effects of obesity in the colon (Jain and Bird 2010; Flores and others 2012; Liu and others 2012), there are several reasons to believe that interleukin-1β (IL-1β) plays an independently prominent role. For example, Tu and others showed that upregulated expression of IL-1β in the gastrointestinal epithelium of mice is, by itself, sufficient to incite inflammation and cancer (Tu and others 2008). Also, in another study IL-1β, expression was found to be necessary for the acquisition of an invasive phenotype and for the stimulation of angiogenesis in several different types of tumors (Allott and Hursting 2015). Moreover, several human studies have observed that polymorphisms in the IL-1β gene that increases its production or activity are independently associated with a higher risk of cancer or a shorter cancer survival (Voronov and others 2003; Tu and others 2008). Also, a prominent role played by IL-1β signaling in carcinogenesis may relate to its multiplier effect, since it upregulates the expression of several other proinflammatory elements such as TNF-α, IL-6, and COX-2 (Howell and others 2003; Voronov and others 2003; Tu and others 2008) and regulates insulin (Gao and others 2014) and leptin secretion (Dinarello and others 2010). The latter 2 molecules are relevant since elevations in each are also postulated to play significant roles in obesity-promoted carcinogenesis (Dinarello 2006).
IL-1β is a promoter of multiple procarcinogenic signaling pathways, including the Wnt cascade. Although basal activity of the Wnt signaling pathway is critical for maintenance of stem cell properties of the colonic crypt (de Lau and others 2007), overactivation of the pathway is a pivotal step in the early development of most CRCs (El-Omar and others 2000). The downstream effects resulting from overactivation of the Wnt signaling cascade on colonocytes include a diverse array of procarcinogenic events, including hyperproliferation in the base of the colonic crypts and movement of actively proliferating cells up the crypt wall (Sansom and others 2004). Thus, we hypothesized that the absence of IL-1 signaling would lead to a reduction in proinflammatory cytokine secretion in the colons of obese mice, coupled with an attenuation in Wnt signaling and a decrease in epithelial proliferation. To examine this hypothesis, we utilized a transgenic mouse lacking a functional IL-1 receptor.
Methods
Animal studies
All animal procedures were approved by the Animal Use and Care Committee of the U.S.D.A. Human Nutrition Research Center at Tufts University. Two strains of male mice were used for this study; wild-type (WT) C57BL6/J and IL1R1tm1lmx (#664 and #3245, respectively; Jackson Laboratories, Bar Harbor, ME). Although the latter animals lack a response to both IL-1α and IL-1β, IL-1β appears to be, by far, the predominant form of IL-1 acting upon nonneoplastic colonocytes in vivo (Barber and others 2000).
At 8 weeks of age, animals were randomized to a 10% low-fat diet (LFD) or 60% high-fat diet (HFD, Research Diets, D12450BM, D12492M, respectively) for 16 weeks; 10 animals were allocated to each group. At diet week 15, body composition was measured by MRI (100H; EchoMRI, Houston, TX). After 16 weeks on diet, mice were euthanized by CO2 asphyxiation followed by cervical dislocation and exsanguination by cardiac puncture. The abdomen was then opened and the large intestine removed and placed onto an ice-cold glass plate. The proximal half of the colon was used to isolate colonocytes. Colonocytes were isolated at 5°C to arrest their metabolism, using a previously described ethylenediaminetetraacetic acid (EDTA) chelation and mechanical agitation method (Schwartz and others 1991), and then stored at −80°C. The edge of a microscope slide was used to scrape mucosa from the distal half of the colon, and mucosa was then stored at −80°C. Blood was spun at 1,000 g and plasma stored at −80°C.
Insulin and leptin
Plasma insulin and leptin values for each animal were measured using an electrochemiluminescent assay, according to the manufacturer's guidelines (K15124C-1, S600; MesoScale, Rockville, MD).
Colonic cytokines
Whole cell lysates were generated from isolated colonocytes and colonic mucosa from using RIPA buffer (#89900; ThermoFisher) to which protease and phosphatase inhibitors (Roche, Indianapolis, IN) were added according to the manufacturer's recommendations. Protein was quantified using the Bradford Assay (Bio-Rad, Hercules, CA). IL-1β, IL-6, TNF-α, and interferon-γ (IFNγ) protein levels were then measured in colonocytes and colonic mucosa by electrochemiluminescence (S600; MesoScale). Cytokine concentrations are adjusted for total protein and reported as picograms per milligram of protein.
Epithelial proliferation
Proliferation was assessed by quantitative immunohistochemistry (qIHC). For qIHC, the paraffin embedded slides were deparaffinized in xylene, followed by rehydration in ethanol. Slides were incubated with Ki-67 antibody in PBS and 1% bovine serum albumin (cat #4501880; Sigma-Aldrich). The antibody was diluted 1:500 in the antibody solution. Following incubation with the biotinylated anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA), the slides were treated with Vectastain Elite ABC reagent (Vector Laboratories) and hematoxylin counterstain. Five or more midlongitudinal sections of crypts from each animal were assessed: only those with the base touching the muscularis mucosa and having an open lumen at the top qualified for inclusion. Quantification was done by a single individual (A.C.P.), blinded to group assignment, under 400-fold magnification. The extent of proliferation zone expansion was assessed by first visually dividing the crypt into upper, middle, and lower thirds. Normally, Ki-67 cells reside exclusively in the bottom third (Tamura and others 2002); the metric used to measure expansion of the proliferation zone was the mean number of Ki-67 positive cells located in the middle and top thirds of the crypt.
Wnt activation
Wnt activation was measured using qIHC for intranuclear β-catenin (primary antibody: #610154; BD Biosciences, San Jose, CA) as previously described (Iwamoto 2000), using the same quality control measures described above for the Ki-67 IHC. β-catenin that remains unphosphorylated at its N-terminal phosphorylation sites is “active” since it is available for nuclear translocation, where it promotes transcription of downstream Wnt signaling genes (de Lau and others 2007).
Statistics
All data are reported as mean ± SEM. Statistical calculations were performed in GraphPad Prism (La Jolla, CA) using a 2-way analysis of variance and Tukey's post hoc test for multiple comparisons when appropriate. Significance was accepted at a 2-tailed P value of <0.05.
Results
Physiology
At the end of the study IL1R−/− and WT mice on the HFD had achieved body weights 36% and 34% greater than their LFD controls, respectively (P < 0.01 for each strain, Fig. 1A). IL1R−/− and WT mice on the HFD also developed 59% and 61% greater fat mass compared to the LFD controls, respectively (P < 0.01; Fig. 1B). IL1R−/− mice on the HFD transiently weighed more than their high-fat fed WT counterparts at week 5 only. Regardless of genotype, mice consuming the HFD diet had significantly elevated plasma insulin (P < 0.05; Fig. 1C) and leptin (P < 0.01; Fig. 1D) concentrations compared with mice on the LFD diet.

HFD: effects on body weight, body composition, and plasma insulin and leptin.
Colonic cytokines
Mucosal concentrations of IL-1β and TNF-α in WT mice consuming the HFD were 76% and 29% greater than in WT LFD mice, respectively (P = 0.04 for both IL-1β and TNF-α; Fig. 2A, B). In contrast, no significant increases in these 2 cytokines were observed in the obese IL1R−/− mice compared with either the WT or IL1R−/− controls. There were no significant differences in colonic IL-6 or IFNγ among the 4 groups.

High-fat-fed WT mice have an elevation in proinflammatory cytokines in the colonic mucosa. Colonic
Cytokine concentrations in isolated colonocytes were similarly compared and no significant differences between the 4 groups were evident for any of the cytokines.
Epithelial proliferation
WT HFD-fed mice displayed 2.3-fold more Ki-67 positive cells in the upper two-thirds of the colonic crypts than their lean counterparts did (P = 0.04; Fig. 3). This increase was not present in the HFD IL1RKO animals. When proliferation was instead measured by the mean percentage of crypt cells that were Ki-67 positive (ie.: the labeling index), there were no significant differences among the groups. However, both rodent (Barnes and others 1999) and human studies (Ponz de Leon and others 1988; Paspatis and others 1998) have shown that expansion of the proliferation zone is a more accurate predictor of subsequent advanced neoplasms than the labeling index.
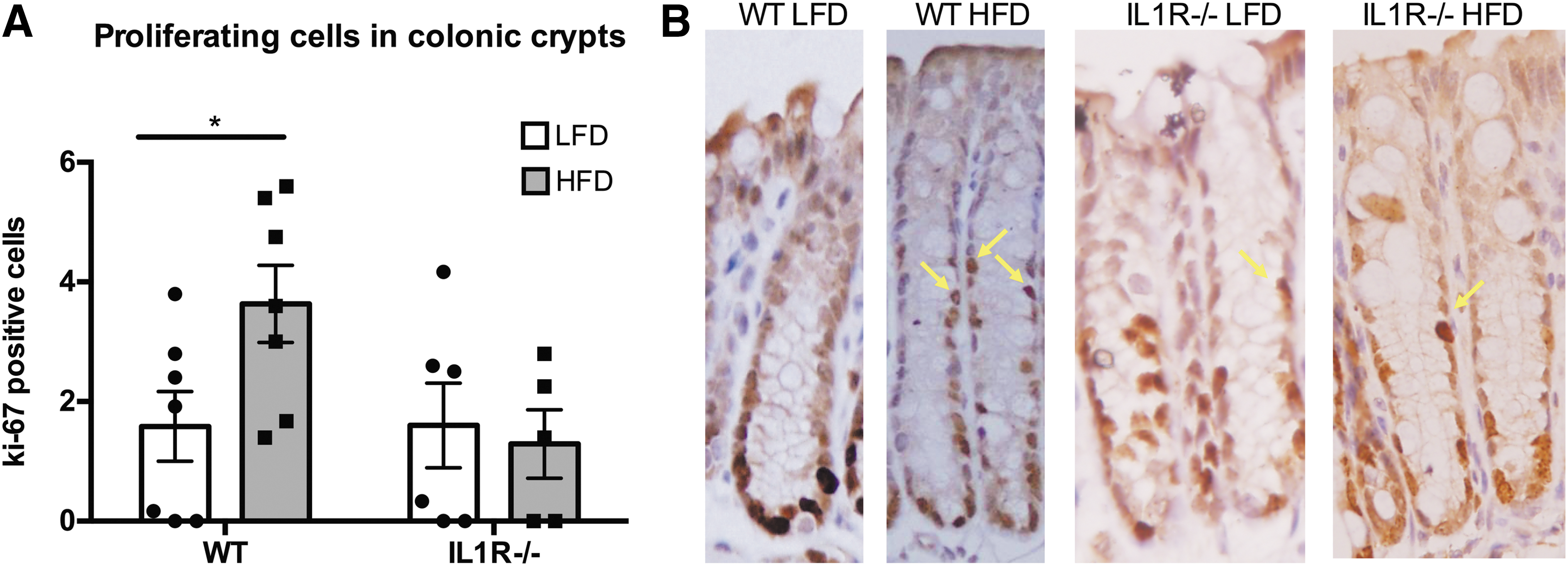
Proliferative cells in the upper two-thirds of colonic crypts.
Wnt signaling
WT mice fed the HFD diet displayed a 1.9-fold greater mean number of colonic crypt cells positive for intranuclear β-catenin than WT mice fed the LFD (2.3 ± 0.4 versus 1.2 ± 0.2 cells/crypt, respectively, P = 0.03) as seen in Fig. 4. In contrast, the number of β-catenin positive cells in the IL1R−/− mice receiving the HFD was not significantly greater compared with the knockout animals receiving the LFD (1.8 ± 0.4 versus 1.3 ± 0.3, respectively, P = 0.30).
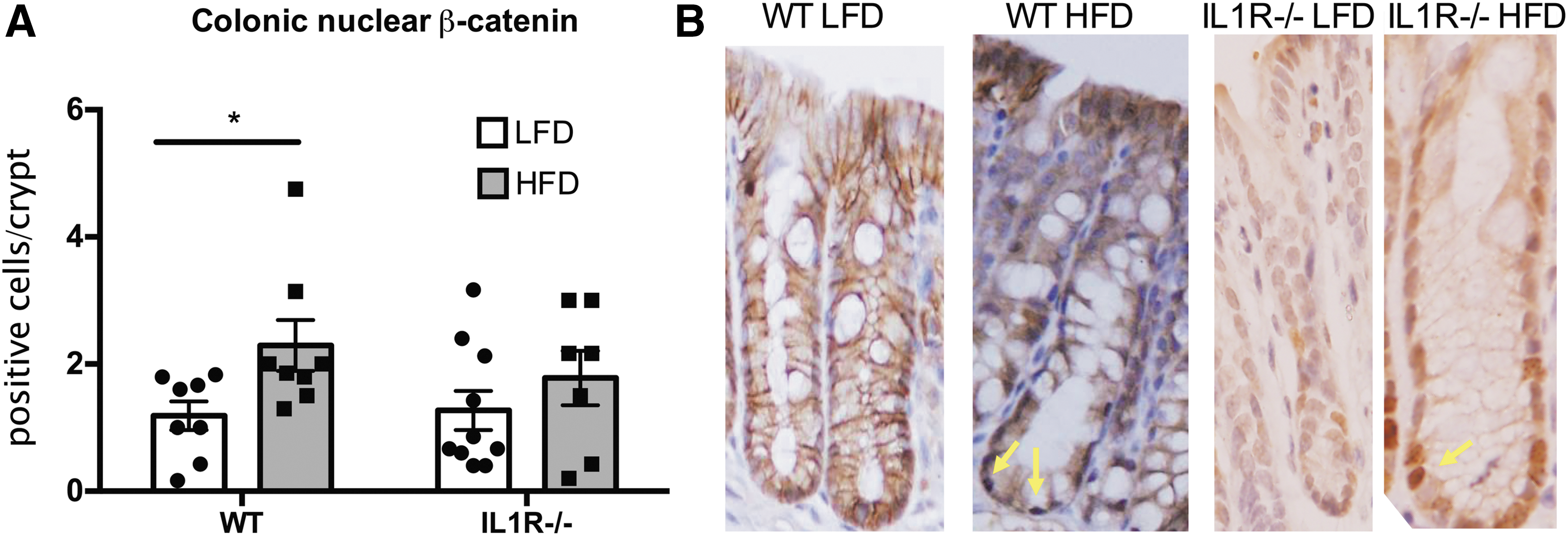
Nuclear β-catenin in colonic crypts. Obesity elevates Wnt signaling in the colon of obese male mice.
Discussion
The link between obesity and CRC has been well documented in epidemiologic studies (Calle and Kaaks 2004; Gunter and Leitzmann 2006), and causality has been proved in animal models (Gravaghi and others 2008; Flores and others 2012; Karim and Huso 2013). Furthermore, there is growing evidence that the inflammatory environment created in the colon by obesity is an important mechanistic factor in this relationship (Jain and Bird 2010; Flores and others 2012; Liu and others 2012). The focus of our study was to provide explicit evidence that the proinflammatory cytokine IL-1β mediates, at least in part, obesity-promoted elevations in colonic cytokines, proliferation, and alterations in procarcinogenic signaling pathways.
Our observations confirm that diet-induced obesity results in an elevation in colonic TNF-α and IL-1β—as has been previously observed (Jain and Bird 2010; Flores and others 2012; Liu and others 2012)—as well as increases in proliferation and Wnt signaling. Very importantly, these protransformational changes depended on the presence of an intact IL-1 signaling system. These effects of IL-1 signaling are consistent with existing literature that indicates that IL-1β produces a positive feedback that magnifies inflammation 10, and that the cytokine stimulates Wnt signaling (Kaler and others 2009, 2009). It is not surprising that elevations in both IL-1β and TNF-α occurred when intact IL-1 signaling was present since there is a considerable evidence that the introduction of IL-1β into biological systems increases the transcription and translation of TNF-α (Gao and others 2014) as well as the production of other proinflammatory molecules (Howell and others 2003). Furthermore, there is a great degree of cross talk among the downstream signaling events incited by TNF-α and IL-1β (Yang and others 2010). The ability of IL-1β to regulate the production and downstream signaling of a host of other proinflammatory cytokines underscores the potential importance of this particular cytokine in inciting events leading to the development of obesity-promoted colon cancer.
Although it is not of primary interest to the focus of this study, it is nevertheless interesting that colonic concentrations of IL-1β and TNF-α rose modestly to an intermediate level even in the knockout animal on a LFD (Fig. 2A, B). Although no conclusive explanation for this emerges from our data, other investigators have observed that the expression of cell-signaling ligands commonly rise when the receptor for those ligands is disabled (Huang and others 2001).
It is notable that the rise in colonic cytokines was only observed in the mucosal scrapings and not in isolated colonocytes. The inflammatory state produced by obesity is thought to be largely due to the secretion of proinflammatory molecules by macrophages (Bain and others 2013; Cassol and others 2015). Macrophage recruitment, and the cytokine secretion that ensues, has consistently been observed in the adipose tissue of obese mice (Dam and others 2016). Thus, colonocytes do not appear to be the source of the elevated colonic cytokine concentrations observed in obesity. Rather, macrophages or other immune cells located in the lamina propria underlying the epithelium are more likely the primary source of the mediators. Future studies will be necessary to conclusively identify the particular cell type(s) that are responsible for these elevations in colonic cytokines.
Rodent studies have demonstrated a significant positive correlation between expansion of the proliferation zone and the development of colonic carcinomas (Barnes and others 1999), and clinical studies have similarly observed that expansion of the proliferation zone is more closely linked to the development of human colonic neoplasms than an increase in the labeling index (Terpstra OT and others 1987; Ponz de Leon and others 1988; Risio and others 1991; Paspatis and others 1998). It is therefore of considerable importance that we detected a significant expansion of the proliferation zone in the colons of obese mice, even though no change in the labeling index was observed. The expansion of the proliferation zone in the obese WT mice was another observation that was absent in the IL1R−/− obese mice (Fig. 3), indicating that IL-1 signaling is an important factor in producing this abnormal pattern of epithelial proliferation that accompanies obesity. The expansion of the proliferation zone that accompanied obesity in this study would be expected since IL-1β also activated Wnt signaling, one downstream effect of which is enhanced proliferation (Kaler and others 2009). The link between IL-1 signaling and proliferation has been identified previously (Vela and others 2002) in various in vitro models, but the observation that IL1R−/− mice are resistant to the proliferative effect of obesity provides further evidence for the particularly pivotal role of this cytokine in the development of CRC.
Collectively, our observations indicate that obesity elevates Wnt signaling in colonocytes and that this effect is lost when IL-1 signaling is ablated (Fig. 4). In normal colonic epithelium, Wnt signaling is critical for sustaining adequate colonocyte turnover, but when inappropriately activated, it is generally regarded as one of the earliest, and most consistent, steps in the molecular carcinogenesis of spontaneous CRCs (Clevers and Nusse 2012). Activated β-catenin is the final common mediator of canonical Wnt signaling and the nuclear availability of this molecule concurs with the magnitude of signaling activation. Since the apparent increase in Wnt activity accompanying obesity that was measured by qIHC was not, by itself, entirely convincing, we sought further evidence of Wnt activation in a follow-up study, which did demonstrate a significant increase in the active form of β-catenin in the colonocytes of obese WT mice (Fig. 4). Numerous investigators have demonstrated that increases in the cellular levels of nonphosphorylated β-catenin concur with increased signaling through the Wnt pathway and its downstream sequalae (van de Wetering and others 2002; van Noort and others 2002). Furthermore, the fact that the obesity-incited elevation in Wnt signaling and accompanying expansion of the proliferation compartment were abolished by ablation of IL-1 signaling, while plasma insulin and leptin were not altered by the loss of IL-1 responsiveness, indicates that the cytokine mediates its effects on Wnt and proliferation through pathways other than its known impact on the insulin (Gao and others 2014) or leptin axes (Dinarello and others 2010). The likely mediator would be Akt, which is activated by IL-1β and in turn activates Wnt signaling (Kaler and others 2009).
This study possesses certain limitations. First, the engineered mice we used lack a response to both IL-1α and IL-1β, and therefore, we cannot definitively distinguish between the effects produced by these 2 forms of IL-1. Nevertheless, IL-1β appears to be, by far, the principal form of IL-1 acting upon nonneoplastic colonocytes (Barber and others 2000), so we postulate that most of the IL-1-mediated effects in this study are due to IL-1β. Obesity can be reliably induced in male mice, but for reasons that remain unclear, a significant proportion of female mice are resistant to the induction of obesity by a HFD, as described by prior investigators (Shi and Clegg 2009). It was for this reason that we chose to confine these studies to male mice. We therefore cannot exclude the possibility that our observations do not apply females.
This study contributes several novel pieces of information about the relationship between IL-1 signaling and early events in obesity-promoted colonic carcinogenesis. First, IL-1 signaling appears to be an important effector of several of the procancerous biochemical, cell signaling, and cytokinetic anomalies that obesity produces in the colon. Second, the discrepancies we observed by comparing results from isolated colonocytes versus those from scraped colonic mucosa suggest that the origin of the excess cytokines is not the colonocytes themselves. These observations underscore the importance of IL-1 signaling in regulating physiologic and molecular mechanisms relevant to the development of obesity-promoted colonic carcinogenesis and, by doing so, offer a signaling pathway that can be targeted to mitigate the increased risk of cancer due to obesity.
Footnotes
Acknowledgments
We thank the staff of the Comparative Biology Unit at the HNRCA for their assistance with animal care, the Boston Nutrition Obesity Research Center for use of their rodent EchoMRI, and Dr. Andrew Greenberg for his comments on the study design. This study was funded, in part, by the Prevent Cancer Foundation and the Agricultural Research Service of the United States Department of Agriculture, Cooperative Agreement #58-1950-0-014. We also thank the Drs. Joan and Peter Cohn Research Award and the Jean Mayer HNRCA Director's Student Innovation Fund for their generous support. Opinions and ideas expressed in this publication are not necessarily those of the U.S. Department of Agriculture.
Author Disclosure Statement
No competing financial interests exist.