
Abstract
Accumulating evidence has shown that atherosclerosis is an inflammatory disease. The pathogenesis of atherosclerosis has been confirmed to involve an imbalance in anti-inflammatory/proinflammatory processes. Interleukin-23 (IL-23) is a heterodimeric cytokine composed of the IL-23p19 and IL-12p40 subunits of the IL-12 family. Experiments show that IL-23 induces CD4+ T cells to differentiate into T helper type 17 cells, promotes the expression of interferon-γ, and inhibits the production of Foxp3. Therefore, IL-23 induces and exacerbates effector T lymphocyte/regulatory T cell imbalance. IL-23 receptor (IL-23R) is expressed not only in T cells but also in dendritic cells (DCs) and macrophages. IL-23R can enhance its antigen-presenting ability through the autocrine pathway, enabling it to infiltrate lesion sites, promote its secretion of a large number of inflammatory factors, and upregulate proinflammatory DCs and macrophages. IL-23 binds IL-23R on the surface of target cells and transmits signals through Janus kinase 2/signal transducer and activator of transcription channels, participating in the occurrence of chronic inflammatory diseases and autoimmune diseases. Therefore, the use of IL-23 or IL-23R is a potential therapeutic approach for treating inflammatory diseases, including atherosclerosis. In this article, we hypothesize that IL-23 may be a novel target for the treatment of atherosclerosis, and further study is needed to determine the precise role of IL-23 in atherosclerosis and the associated signaling pathways.
Introduction
V
Cytokines may be roughly divided into two categories according to their modulatory role in atherosclerosis. Proinflammatory cytokines, effector T lymphocytes (Teffs) [such as T helper type 1 (Th1) and T helper type 17 (Th17) cells], mature dendritic cells (mDCs), M1 macrophages, and inflammatory factors [such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-17 (IL-17)] exert proatherogenic effects. The Th1 cell subset is characterized by IFN-γ production and T-bet expression, whereas the Th17 cell lineage produces IL-17 (Mallat and others 2009). However, anti-inflammatory cytokines, regulatory T cells (Tregs), immature DCs (secrete IL-10), and M2 macrophages can help ameliorate atherosclerosis (Wu and others 2013).
IL-23 is a newly discovered cytokine of the IL-12 family composed of IL-12 and IL-35. The p19 subunit associates with the p40 subunit to form IL-23, IL-12 (IL-12p35/p40), and IL-35 (IL-12p35/Ebi3) (Yan and others 2017). IL-23 is a heterodimer of the nonglycosylated protein IL-23p19 and the glycosylated protein p40 (Floss and others 2015). IL-23 is primarily secreted by activated macrophages and DCs. IL-23 is regarded as a crucial cytokine for the pathogenesis of autoimmune diseases and the circulatory system. Evidence shows that IL-23 is essential for the secretion of IL-17 family cytokines by Th17 cells (Lubberts 2015). This cytokine also promotes the expression of IFN-γ and IL-22 (Belladonna and others 2006). IL-23 has been evaluated in clinical studies of autoimmune diseases and the circulatory system. However, research on IL-23 in the field of atherosclerosis has just begun. Although several animal and clinical studies have shown that IL-23 is involved in the process of atherosclerosis, its precise effects and mechanisms remain unclear.
Biological Characteristics of IL-23
IL-23 is a heterodimeric cytokine composed of the p19 (a four-helix bundle cytokine, 19,000 Da molecular weight) and p40 (a soluble class I cytokine receptor p40, 40,000 Da molecular weight) subunits that are connected with a disulfide bond (Yan and others 2017). IL-23 is activated to participate in innate and adaptive immune responses through the IL-23 receptor (IL-23R) and the shared receptor IL-12Rβ1 (Floss and others 2013). IL-23R and IL-12Rβ1 are glycosylated type I membrane proteins (Floss and others 2015). Macrophages, DCs, lymphocytes, and endothelial cells can all secrete IL-23. Macrophages and DCs are the main sources of IL-23, but IL-23 is an important marker of type M1 macrophages as well. The most prominent role of IL-23 is to regulate the activity of T cells, as IL-23R is mainly expressed on the surface of T cells. Studies have shown that IL-23 induces the differentiation of CD4+ T lymphocytes into Th17 cells; therefore, IL-23 is not only a necessary factor for the pathogenesis of Th17 cells, it also plays an important role in autoimmune and inflammatory diseases (Croxford and others 2014; Lubberts 2015; Teng and others 2015).
IL-23 binds to IL-23R and IL-12Rβ1 on the surface of target cells, thus activating the corresponding signaling pathway and promoting intracellular signal transduction, with a variety of biological effects. IL-23 has been shown to promote the expression of IFN-γ and IL-22 (Belladonna and others 2006). Izcue and others (2008) demonstrated that endogenous IL-23 inhibits the expression of Foxp3 that is a key transcription factor in the Treg-mediated maintenance of immune homeostasis. Thus, IL-23 induces and aggravates the Teff/Treg imbalance and upregulates proinflammatory cytokines and the secretion of inflammatory factors to enhance inflammatory responses. IL-23 can also promote the transformation of DCs and macrophages into mDCs and M1 macrophages, respectively (Bastos and others 2005).
Stimulation of the IL-23R complex activates Janus kinase 2 (JAK2) and tyrosine kinase 2 (Tyk2), resulting in phosphorylation of the receptor complex and activation of docking sites for signal transducer and activator of transcription (STAT) 1, 3, 4 and 5; among these molecules, STAT3 is primarily activated. Studies have shown that the phosphorylation of STAT4 increases the secretion of IFN-γ and subsequent differentiation of Th1 cells, whereas the phosphorylation of STAT3 is absolutely necessary for guiding the differentiation of Th17 cells (Tan and others 2009). Studies have predicted the binding sites of IL-23R to murine and human STAT3; these sites are Tyr-504/Tyr-626 and Tyr-484/Tyr-611, respectively (Parham and others 2002). IL-23 shares the same JAK-STAT signal molecules as IL-12, that is, JAK2, Tyk2, and STAT1, 3, 4, and 5, but data have shown that STAT4 activation is notably weaker in response to IL-23. Increasing evidence has revealed that the ability of cells to respond to IL-23 or IL-12 signals is associated with the expression level of IL-23R or IL-12Rβ1, respectively (Floss and others 2013) (Fig. 1).
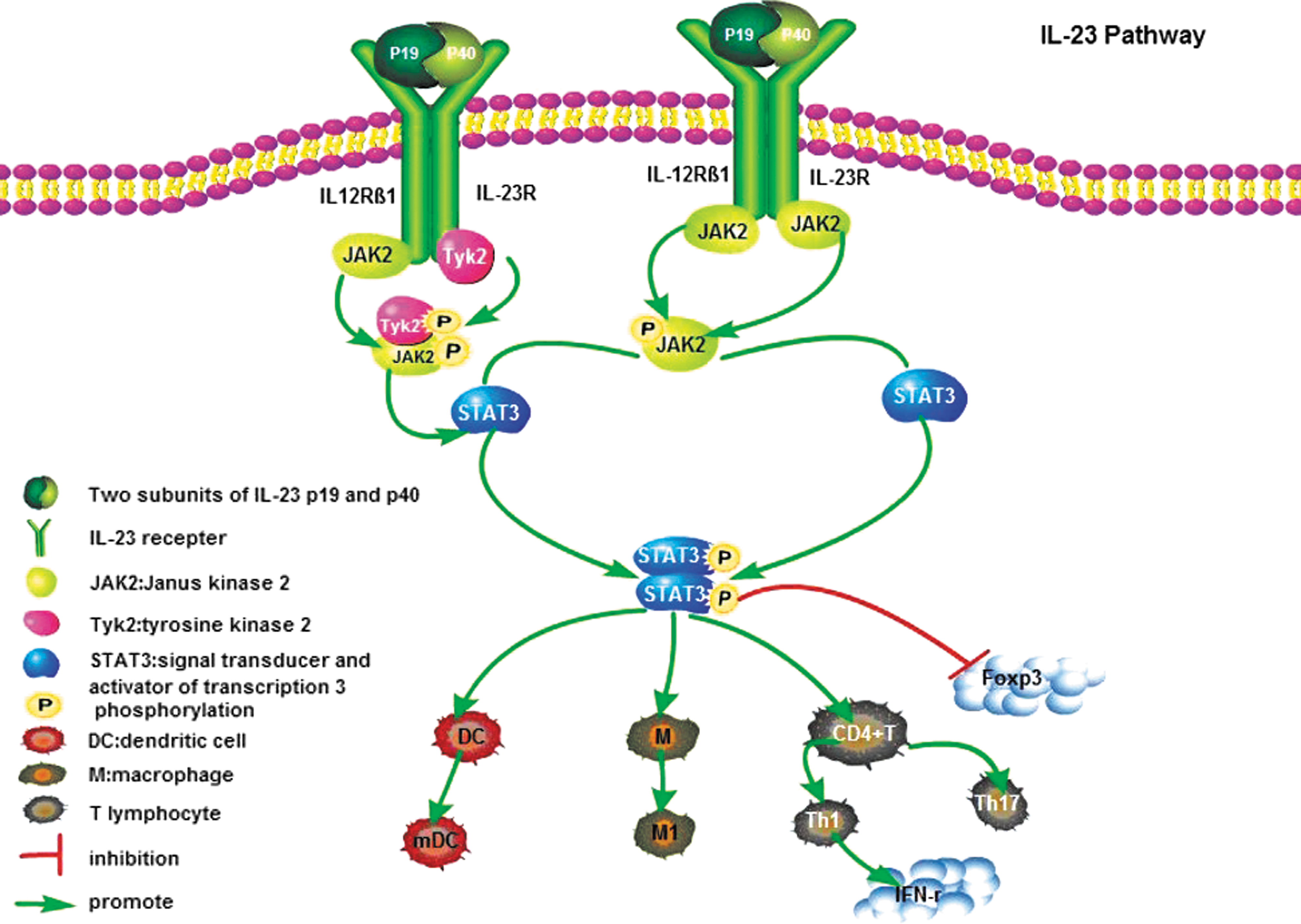
The binding of IL-23 and IL-23R leads to the conformational change of the receptor that activates JAK2 and Tyk2 and promotes the phosphorylation of tyrosine residues, thereby promoting the corresponding phosphorylation STAT factors. Finally, the activated STAT protein dissociates from the receptor, forming a dimer and then entering the nucleus to exert its biological effects. DC, dendritic cell; IFN-γ, interferon-γ; JAK2, Janus kinase 2; M, macrophage; M1, M1 macrophage; mDC, mature dendritic cell; STATs, signal transducer and activator of transcriptions; Th17, T helper type 17; Tyk2, tyrosine kinase 2.
IFNs are divided into type I and type II IFNs, and type I is further divided into two types: IFN-α and IFN-β, whereas type II is IFN-γ. Different types of IFNs originate from different cells, with IFN-α mainly being derived from mononuclear macrophages, whereas IFN-β comes from fibroblasts; furthermore, IFN-γ mainly originates from αβ T cells, γδ T cells, and natural killer cells. IFNs bind to receptors in an autocrine and paracrine manner and produce a series of signaling cascades through receptor-mediated JAK-STAT signaling pathways that exert biological effects (Diebold and others 2003). Studies have shown that the proinflammatory cytokine IFN-γ can induce elevated STAT protein expression (Shea-Donohue and others 2010), and IL-23 can also induce the secretion of IFN-γ through the JAK-STAT signaling pathway.
IL-23 and Immune System Diseases
Recent studies have shown that IL-23 combined with IL-23R promotes intracellular signal transduction and exerts proinflammatory effects. The current review focuses on the role of IL-23 in chronic inflammatory and autoimmune diseases. Studies have shown that psoriasis is associated with atherosclerosis. The clinical symptoms in patients with severe psoriasis are improved after anti-IL-12/23 treatment, but the anti-IL-12/23 treatment may not be effective for treating cardiovascular disease in patients with psoriasis (Ji-Chen and others 2016). Other works suggest that IL-23 blockers might be the optimal treatment for psoriatic patients (Bachelez 2017; Křížová and others 2017). IL-23 may be involved in the pathogenesis and progression of scleroderma by reducing the expression of type I collagen protein and participating in the regulation of fibrosis (Nakayama and others 2017). Feldmeyer reported that in pityriasis rubra pilaris (PRP) patients, lesional skin samples were characterized by a large number of proinflammatory cytokines, including TNF-α, IL-6, IL-12, IL-23, and IL-1β. However, the clinical symptoms of all patients with PRP treated with ustekinumab [an IL-23 Th17 cell pathway blocker] were found to be improved (Feldmeyer and others 2017). A meta-analysis showed a significant association between IL-23R gene polymorphisms and ulcerative colitis (Peng and others 2017). The role of IL-23 in chronic inflammatory and autoimmune diseases and related mechanisms has also been studied in depth.
These data suggest that IL-23 has strong immune regulatory and proinflammatory effects. In addition, the anti-IL-23 monoclonal antibody guselkumab has been used for the treatment of moderate-to-severe plaque psoriasis. The study showed that guselkumab presents more robust efficacy in relation to the area of psoriasis and severity index than did adalimumab (TNF monoclonal antibody) (Gordon and others 2015).
IL-23 and Cardiovascular and Circulatory Diseases
Muhammad conducted a prospective study of 80 patients with aneurysmal subarachnoid hemorrhage (aSAH). The results showed that the serum levels of IL-23 and IL-17 were increased and that aSAH upregulated the IL-23/IL-17 inflammatory axis (Chaudhry others 2017). In another study, three different types of mice were used in an analysis of myocarditis: IL-12p40−/− mice, IL-12p35−/− mice, and mice treated with a neutralizing IL-23 antibody. The experiments demonstrated that IL-23 was involved in the course of myocarditis, rather than IL-12 (Sonderegger and others 2006).
The effects of IL-23 on ischemic cardiac scar formation have also been studied. In a model of myocardial infarction (MI) established in wild-type mice and IL-23p19 knockout mice, IFN-γ/Th1 reactivity was shown to increase with IL-23 deficiency and led to increased myocardial inflammation, reduced cardiac fibroblast activation, early left ventricular enlargement, and a 40% increase in cardiac rupture 30 days after MI (Savvatis and others 2014). Potential associations between the IL-23/IL-17A axis and left ventricular remodeling in the late stage of MI have also been studied in an MI model of coronary ligation (Yan and others 2012). In addition, data from an ischemia/reperfusion (I/R) model suggested that IL-23 could promote myocardial I/R injury by increasing the inflammatory response and oxidative stress (Hu and others 2016).
IL-23 and Atherosclerosis
IL-23 has entered the clinical stage of transformation in the field of chronic inflammatory and autoimmune diseases, and research in the field of atherosclerosis lags markedly behind. A number of clinical studies have indicated a significant increase in IL-23 levels in patients with atherosclerosis (Liu and others 2014; Abbas and others 2015), but the most valuable clinical research published by Abbas and others (2015) found a significant increase of IL-23/IL-23R in carotid plaques. IL-23 markedly increased TNF release in peripheral blood mononuclear cells when costimulated with lipopolysaccharide that was only seen in cells from patients with carotid plaques. In this study, the authors also observed colocalization of IL-23/IL-23R and macrophages within atherosclerotic lesions, suggesting there was an interplay between them. Thus, IL-23 has potent inflammatory effects in monocytes from patients with carotid atherosclerosis. This experiment also indicated that IL-23 increased IL-17 release in monocytes and particularly in peripheral blood mononuclear cells from patients with carotid atherosclerosis. Another study involving apoE/IL-18 double-knockout mice showed that the formation of atherosclerotic plaques increased the expression of Th17 and the numbers of IL-23-producing vascular smooth muscle cells and macrophages, and a thin fibrous cap in lesions caused plaques to break easily (Chen and others 2010). However, the role of IL-23 in atherosclerosis development remains controversial.
Studies have shown that IL-17 can reduce macrophage expression in human carotid artery lesions, increase smooth muscle cell content, and promote plaque stability (Liuzzo and others 2013). Granulocyte macrophage colony-stimulating factor (GM-CSF, Csf2) is a growth factor of myeloid-lineage cells. Studies have shown that GM-CSF is involved in the pathogenesis of atherosclerosis and other chronic inflammatory diseases. For the first time, an animal study has suggested that, as a downstream molecule of GM-CSF, IL-23 plays an important role in GM-CSF-promoted atherosclerosis and induces the release of reactive oxygen species in macrophages. The GM-CSF/IL-23 axis can promote the rupture of atherosclerotic plaques by enhancing the apoptosis of macrophages and DCs (Döring 2015). Subramanian and others (2015) found that Csf2−/−Ldlr−/− mice fed a Western-type diet for 12 weeks were characterized by decreases in the Il-23 messenger RNA level. In that study, the aortic root lesions of these Csf2−/− mice were found to show substantial decreases in the number of apoptotic cells, plaque necrosis, and oxidative stress compared with lesions of control Ldlr−/− mice. Studies have confirmed that the dominant effect of GM-CSF in Ldlr−/− mice is to secrete IL-23 through the activation of macrophages and DCs in atherosclerotic lesions and that IL-23 enhances apoptosis by downregulating the antiapoptotic protein B-cell lymphoma 2, increasing oxidative stress, and promoting the formation of advanced atherosclerotic plaques.
Multiple reports have revealed elevated levels of IL-23 in patients and animals with atherosclerosis, suggesting that IL-23 may have proinflammatory and proatherogenic effects. However, Koltsova and others (2016) identified a novel role for the cytokine IL-23 in the development of atherosclerosis: IL-23 may actually act as an immunoregulatory agent in the presence of hypercholesterolemia, thereby preventing atherosclerosis. This effect may occur because IL-23 plays a different role in the development of atherosclerotic plaques at different times. It may also be that IL-23 plays different roles in humans and animals. Hence, the question arises regarding the type of role IL-23 plays in atherosclerosis. Our research group has also carried out a study in this arena.
Our previous research confirmed that IL-23 expression was significantly increased in an atherosclerosis model induced by hyperlipidemia alone or in an atherosclerosis model induced by both angiotensin II and hyperlipidemia (Kai and others 2015; Ji and others 2017). These findings have led to great interest in the role of IL-23 in the atherosclerosis process. In pilot experiments, we used an anti-IL-23p19 antibody to abolish the IL-23 effect and observe the role of IL-23 in the atherosclerosis model. The results showed that the neutralization of IL-23 not only inhibited the Th17 immune response but also significantly inhibited the Th1 immune response and significantly alleviated atherosclerosis. This study explored the effects of IL-23 as a source molecule on the atherosclerosis process (data not published). Numerous studies have shown that a variety of cells, such as monocytes, macrophages, T lymphocytes, DCs, and endothelial cells, and a large number of cytokines are involved in the chronic inflammatory process of atherosclerosis (Ross 1999; Hansson and Libby 2006; Döring 2015). Imbalances in immune regulation are the root cause of chronic inflammatory disease. Predominant proinflammatory activity can promote the development of atherosclerosis, plaque instability, and emergence of clinical symptoms. In contrast, anti-inflammatory activity can delay atherosclerosis progression, stabilize plaques, and relieve clinical symptoms (Fredman and Tabas 2017). However, the mechanisms underlying the changes in endogenous IL-23 expression and regulation of atherosclerosis remain unclear.
Hypothesis
A Teff/Treg imbalance and polarization of the direction of macrophages and DCs dominate the process of atherosclerosis, and IL-23 can promote these effects. Considering the most recent research studies and the preliminary work of our research group (Ji and others 2009; Lin and others 2013; Kai and others 2015), we hypothesize that IL-23 can regulate atherosclerosis. To elucidate the biological effects of IL-23 on the development of atherosclerosis, we propose the use of IL-23 as an intervention target to correct proinflammatory/anti-inflammatory imbalances and suggest a new strategy for the prevention and treatment of atherosclerosis. However, this hypothesis remains to be tested in animal and cell experiments.
Footnotes
Acknowledgments
The authors thank their mentors Dr. Chang Chao and Dr. Ji Qingwei for their patience in mentoring me. They also thank our seniors, classmates, and teachers for their support of my research.
Author Disclosure Statement
All the authors declare that they have no competing interests.