
Abstract
Interleukin-33 (IL-33) is one of the members of the IL-1 family of cytokines and a ligand of ST2 and IL-1 receptor accessory protein (IL-1RAcP) that is known to affect Th2 inflammatory response with partial effects on Th1 responses. This cytokine is released by epithelial and smooth muscle cells of the airway system during their injury by several environmental stimuli, such as allergens, viruses, helminths, and pollutants. IL-33 is an alarmin that acts as an endogenous danger signal, and it has been known to affect various types of cells, such as mast cells, basophils, eosinophils, T cells, and specific subsets of innate lymphoid cells (ILCs). In recent findings, this cytokine is believed to have a critical role in several types of cancers, such as lung cancer, liver cancer, and head and neck squamous cell cancer. The expression of IL-33/ST2 in cancer tissues shows a close association with tumor growth and tumor progression in several types of cancer, suggesting the IL-33/ST2 pathway as a potential target for therapy.
Interleukin-33, an IL-1 Family Cytokine with Different Manners of Activation
Interleukin-33 (IL-33) is a ligand of ST2 (also known as IL-1R4, IL-1RL1, T1, Der4, and fit-1) (Schmitz and others 2005). This cytokine is produced by various types of immune cells, such as mast cells, macrophages, and dendritic cells, and also by nonimmune cells, such as endothelial cells, epithelial cells, smooth muscle cells, and fibroblasts (Oboki and others 2010).
This cytokine is identical to the previously reported DVS27 and the nuclear factor of high endothelial venules and is increased in vasospastic cerebral arteries in the presence of subarachnoid hemorrhage (Onda and others 1999; Baekkevold and others 2003). It was revealed that IL-33 is associated with heterochromatin via a helix-turn-helix motif of the N-terminal part and acts as a transcriptional repressor in vitro (Carriere and others 2007; Roussel and others 2008). Furthermore, IL-33 also serves the function of a nuclear factor-like IL-1α and HMGB1 (Werman and others 2004; Andersson and Tracey 2011).
The IL-1 family cytokines such as IL-1β and IL-18 are absent from hydrophobic signal peptides for protein secretion, which keeps the precursor proteins in an inactive form in the cytoplasm (Dinarello 1998, 2009). Following the activation of caspase-1 (also known as IL-1-converting enzyme) by inflammasomes, IL-1β and IL-18 precursors mature by cleavage to be secreted through a unique mechanism (Dinarello 1998, 2009). When IL-33 was first identified, it was considered that IL-33 experiences an analogous process of other IL-1 family cytokines to be mature and secreted (Schmitz and others 2005). In the case of this cytokine, however, even the full-length protein without maturation has a biological function (Fig. 1A) (Cayrol and Girard 2009; Luthi and others 2009; Talabot-Ayer and others 2009). Also, the caspase-mediated proteolysis that occurs during apoptosis and activation of inflammasomes is not necessary for activation or secretion of IL-33.
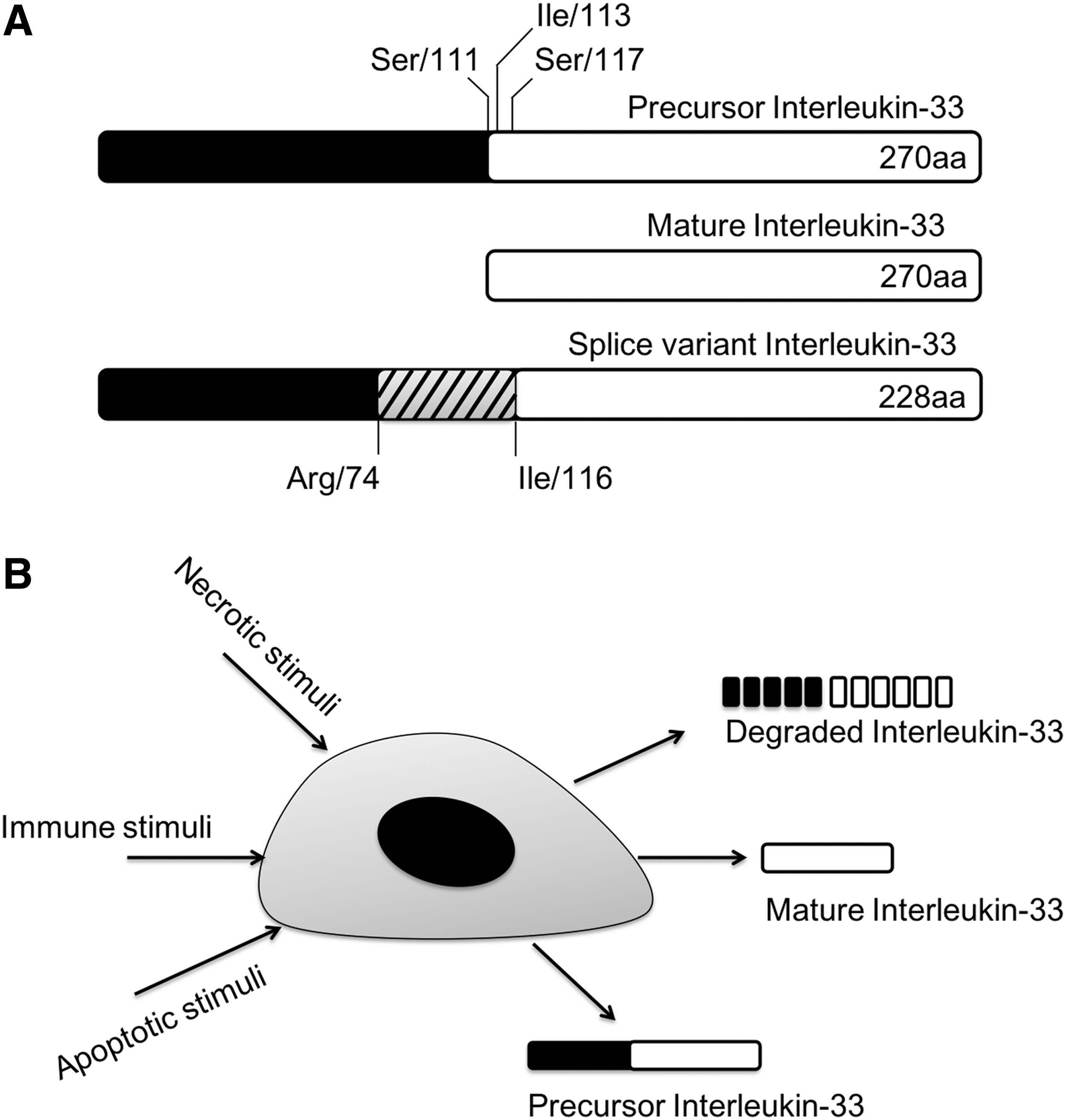
The activation of precursor IL-33.
In another study on IL-33, the cleavage of pro-IL-33 in apoptotic cells leads to inactivation of IL-33, meanwhile full-length pro-IL-33 proteins released from damaged cells are active (Cayrol and Girard 2009; Luthi and others 2009; Ohno and others 2009). These authors reported that the caspase-1 cleavage site is not the initially proposed Asp110-Ser111 but Asp178-Gly179 of IL-33 protein, which is a consensus sequence of caspase-3 cleavage. Caspase-3 and -7 cleaves Asp178-Gly179 site, which destroys the biological function of pro-IL-33 and leads to apoptosis (Schroder and Kaufman 2005; Cayrol and Girard 2009; Luthi and others 2009; Hong and others 2011).
A study showed calpain-dependent processing of pro-IL-33 in the similar manner of IL-1α. Calpain cleavage processes IL-33 maturation and releases this cytokine from human epithelial and endothelial cells. Nevertheless, the functional consequence of this cleavage is still unclear (Hayakawa and others 2009). In human macrophages and mast cells, caspase-1/8 and calpain are unessential for the secretion of IL-33 (Ohno and others 2009). Peritoneal macrophages of caspase-1 knockout mice released IL-33, and stimulation augments the secretion with lipopolysaccharide (LPS) or phorbol 12-myristate 13-acetate plus ionomycin (Ohno and others 2009). These 2 studies show conflicting results on cleavage and secretion of IL-33. The study of calpain-dependent IL-33 maturation was performed in cells originated from humans, whereas Ohno and others (2009) revealed the process of IL-33 with mouse peritoneal neutrophils. The homology of human and mouse IL-33 sequence is significant; however, in amino acid level, the identity is 55%.
Studies have identified splice variants of IL-33. One of the splice variants of IL-33 that lacks exon 3 was revealed to be more active than the full-length form of IL-33 (Hong and others 2011). The cleaved form of IL-33 by neutrophil elastase, cathepsin G, or PR3 shows 10-fold higher activity than that of full-length IL-33 (Bae and others 2012; Lefrancais and others 2012). Fascinatingly, PR3 has functions of both activation and inactivation of precursor IL-33. PR3 cleaves the precursor IL-33 at the N-terminus, and the mature IL-33 is highly active and sufficient to induce inflammatory cytokines (Bae and others 2012). However, prolonged incubation of IL-33 with PR3 led to degradation of IL-33 (Fig. 1B) (Bae and others 2012). IL-33 has a consensus PR3 cleavage motif as ABZ-X3X2X1-ANB-NH2. Cleavage at this site by PR3 activates the cytokine (Wysocka and others 2008; Bae and others 2012). The full mapping of the IL-33 fragments by the cleavage of PR3 is not known yet since PR3 cleaves PR3 at specific sites first but chops up IL-33 after all (Bae and others 2012).
There is a report that IL-33 can be leaked from damaged cells instead of other classic secretion mechanisms of usual cytokines. In their findings, IL-33 works as a damage-associated molecular pattern molecule or as an alarmin like IL-1α and HMGB1 that are leaked from necrotic cells following tissue damage (Moussion and others 2008).
IL-33 Receptor
The IL-33 receptor (IL-33R) is a heterodimeric complex of ST2 and IL-1 receptor accessory protein (IL-1RAcP) (Ali and others 2007; Chackerian and others 2007; Palmer and others 2008). ST2 was known as an orphan receptor for >10 years before the discovery of IL-33 (Trajkovic and others 2004). Over the years, ST2 has been shown to be a selective marker for murine and humans (Trajkovic and others 2004). This molecule is expressed in mast cells (Allakhverdi and others 2007), eosinophils (Cherry and others 2008; Suzukawa and others 2008b), and even in basophils (Suzukawa and others 2008a) without known ligands. These cells produce inflammatory cytokines and chemokines IL-4, IL-5, IL-6, IL-13, and IL-8 in response to IL-33 (Allakhverdi and others 2007; Iikura and others 2007; Cherry and others 2008; Pecaric-Petkovic and others 2009). In a recent study, circulating CD34+ hematopoietic progenitors respond to IL-33 by expressing ST2 and releasing high levels of Th2-associated cytokines (Allakhverdi and others 2009). Such observations suggest what the potential role of IL-33 is in Th2 immune responses and the relevance to allergic inflammatory diseases, such as asthma and atopic dermatitis.
Two splice variants of ST2 have been reported: transmembranous ST2L and soluble sST2. The alternative splicing is regulated under the control of 2 distinct promoters of ST2 DNA (Oboki and others 2010; Smith 2010; Ohno and others 2012). The transmembrane form, ST2L, is the functional component of IL-33R, whereas the soluble form, sST2, is the decoy receptor of IL-33 in the similar manner of IL-1 receptor antagonist (Oboki and others 2010; Smith 2010; Ohno and others 2012). In humans, a third variant, ST2V, is also reported from the gastrointestinal tract, but the function is still unclear (Tago and others 2001).
IL-1RAcP is a co-receptor for IL-33 signaling, and this molecule acts as a shared co-receptor for other IL-1 family members (Ali and others 2007; Chackerian and others 2007; Palmer and others 2008). IL-1RAcP forms a complex with ST2L in a ligand-dependent manner, and the affinity of mouse IL-33 for ST2L is increased in complex with IL-1RAcP (Palmer and others 2008). The action of IL-33 requires IL-1RAcP, and this can be experimentally inhibited by dominant-negative IL-1RAcP mutants or by anti-IL-1RAcP antibodies. It is also known that sST2 and soluble IL-1RAcP, a splice variant of IL-1RAcP, inhibit the activity of IL-33 synergistically (Palmer and others 2008).
IL-33 Signaling
In mast cells of the mouse, IL-33 binds to ST2L and recruits IL-1RAcP, which induces the activation of common signaling pathways of the Toll/interleukin-1 receptor (TIR) family. This leads to the recruitment of MyD88 and the activation of ERK, JNK, p38 MAPK, and NF-κB pathways (Schmitz and others 2005; Ho and others 2007; Baumann and others 2015). These pathways were also confirmed in other types of cells, such as basophils, eosinophils, epithelial cells, endothelial cells, myocytes, and monocytes (Pecaric-Petkovic and others 2009; Chow and others 2010; Yagami and others 2010; Yndestad and others 2010). Although IL-33 activates NF-κB in cardiomyocytes by itself, IL-33 reduces angiotensin II and phenylephrine-induced NF-κB activity, which infers that IL-33 signal is dependent on cellular environment (Sanada and others 2007).
It was reported that the activation of NF-κB, p38, and JNK signaling was dependent on TNF receptor-associated factor 6 (TRAF6), whereas ERK activation is TRAF6 independent from ST2L-overexpressed fibroblasts (Funakoshi-Tago and others 2008). Besides, the activation of NF-κB was Janus kinase 2-dependent unlike ERK, p38, or JNK in the ST2L-overexpressed fibroblasts (Funakoshi-Tago and others 2011). Thus, SIGIRR (single Ig IL-1-related receptor) has a negative regulation on the IL-33-ST2L signaling pathway, which inhibits the activation of ERK, JNK, and NF-κB like other TIR family members (Bulek and others 2009).
It has also been reported that IL-33 crosstalks with c-kit, which is related to the activation of PKB, ERK, JNK, and STAT3 in mast cells, unlike classical TIR signals. This induces the optimal cellular responses to IL-33 (Drube and others 2010). Thus, IL-33 activates GRB2-associated-binding protein 2 (GAB2), phospholipase C-γ2, and Syk during the differentiation of osteoclasts. This means that IL-33 and immunoreceptor tyrosine-based activation motif (ITAM)-bearing receptors crosstalk potentially (Mun and others 2010). Overall, these findings are relevant to the cross talk between receptor activator of NF-κB ligand (RANKL) and signaling through the ITAM in Fc receptor-γ or DAP12 during osteoclastogenesis and of other directions of TLR-ITAM cross talk (Takayanagi 2007; Ivashkiv 2008; Palmer and Gabay 2011; Baumann and others 2015).
However, 1 caveat is that most data related to IL-33 signaling via ST2L were conducted using recombinant 18 kDa IL-33 that is now believed to be an artificially truncated form of IL-33. Although the full-length IL-33 binds to ST2L, recruits IL-1RAcP, and activates NF-κB (Ali and others 2007; Cayrol and Girard 2009; Luthi and others 2009; Talabot-Ayer and others 2009), the 18 kDa IL-33 and full-length IL-33 have differences in specific activities (Luthi and others 2009; Smith 2011).
Cellular Targets of IL-33
IL-33 is released from damaged structural cells and activates mast cells. IL-33 binds to ST2L on either mature or precursor mast cells (Allakhverdi and others 2007) and starts subsequent activation of NF-κB and transcription of proinflammatory cytokines, such as IL-1β, IL-6, IL-13, TNF-α, chemokines, and prostaglandins (Ali and others 2007; Haenuki and others 2012; Ohno and others 2012). IL-33 induces cytokine production in the presence or absence of co-stimulation of mast cells via IgE/antigen–FɛRI signals (Iikura and others 2007; Haenuki and others 2012). IL-33 primes mast cells for activation by IgG immune complexes and increases their survival and adhesion (Kaieda and others 2012).
IL-33/ST2L interactions induce degranulation in response to IgE cross-linking stimuli, and it enhances basophil migration to eotaxin without effect on CCR3 expression (Chan and others 2001; Smithgall and others 2008). Thus, IL-33 synergizes with IL-3 to induce IL-4 expression and CD11b production by basophils, which enhance basophil adhesiveness (Smithgall and others 2008). Like mast cells, basophils also produce proinflammatory cytokines, such as IL-1β, IL-4, IL-5, IL-6, IL-13, and GM-CSF (Smithgall and others 2008; Suzukawa and others 2008a; Pecaric-Petkovic and others 2009).
IL-33 regulates blood eosinophil infiltration in airway inflammation in mice (Stolarski and others 2010). IL-33 stimulates IL-5-dependent eosinophil differentiation from CD117+ progenitors. In human eosinophils, IL-33 mediates survival, upregulates ICAM-1 on the cell surface, and suppresses ICAM-3 and selectin. IL-33 induces the release of proinflammatory cytokine IL-6 and chemokines IL-8 and CCL2 and promotes Siglec-8-mediated apoptosis of eosinophils (Chow and others 2010; Na and others 2012). When mice are treated with IL-33, CCR3, and CC4, their ligands, CCL11, CCL17, and CCL22, increase. These results implicate eosinophil chemotaxis in lung tissue (Louten and others 2011). In IL-33 null mice, eosinophil infiltration and cytokine production were decreased (Louten and others 2011). IL-33 stimulates eosinophils to produce superoxide and degranulation (Cherry and others 2008). In humans, the correlation between blood and pulmonary eosinophilia with elevated IL-33 serum level was reported (Kim and others 2010). Macrophages and dendritic cells also express ST2 but in low levels. ST2 stimulation in dendritic cells increases IL-4, IL-5, IL-13, CCL17, TNF-α, and IL-1β (Su and others 2013). In macrophages, IL-33 amplifies the expression of M2 markers (Espinassous and others 2009). Furthermore, IL-33/ST2 signaling enhances the activation of components of LPS receptor, TLR4, and MD2 (Joshi and others 2010).
It has been known that IL-33 induces Th2 cytokines and chemotaxis of in vitro polarized Th2 cells (Schmitz and others 2005; Komai-Koma and others 2007; Ohno and others 2012). In mouse respiratory system, antigen-specific Th2 cells stimulated by IL-33 induce IL-5 and IL-13, but not IL-4, and these cells are called atypical Th2 cells (Kurowska-Stolarska and others 2008). This was also observed from BAL fluid that IL-5 and IL-13 levels were increased while IL-4 did not change following intranasal administration of IL-33, which suggests that IL-33 is involved in IL-4-independent Th2 cell differentiation (Louten and others 2011). IL-33 induces IL-5 and IL-13 and impressively interferon-γ (IFN-γ), a Th1 cytokine. It also enhances Th2 cytokines in vitro among skewed cells in HDM-specific T cell culture (Smithgall and others 2008). IL-33 is involved in Th2 responses to allergens such as house dust mites, peanuts in experimental animal models of asthma, and food allergy (Chu and others 2013). Interestingly, ST2 or IL-33 null mice have normal levels of Th2 differentiation (Hoshino and others 1999; Townsend and others 2000). The expression level of ST2 is low in CD8 T cells; however, IL-33 enhances the antitumor activity of CD8 T cells (Gao and others 2015). Furthermore, loss of ST2 impaired response to LCMV infection in CD8 T cells (Bonilla and others 2012). iNKT cells have membranous ST2 and express both Th1 and Th2 cytokines under stimulation of IL-33 (Smithgall and others 2008). These results suggest that IL-33 promotes Th2 cytokines in a particular condition as well as Th1 cytokines. NK cells express ST2, and the combination of IL-33 and IL-12 increases IFN-γ levels (Smithgall and others 2008; Bourgeois and others 2009). These data have yet to translate to human disease. These phenomena imply possible existence of another component that supports IL-33R complex, which transduces Th1 signal following the stimulation by IL-33. Recent studies show that T cell subset is not the only type of lymphoid cells with ST2. B1 B cells express ST2, which enhances proliferation capacity, and IgM, IL-5, and IL-13 production (Komai-Koma and others 2011).
In recent studies, various IL-5 and IL-13 producing Lin− c-kit+ Sca-1+ innate lymphoid cells (ILCs) have been reported as a distinct subset of cells from lymphoid progenitors, lymphoid tissue inducer cells, and RORγt+ ILCs (Moro and others 2010; Neill and others 2010; Price and others 2010; Saenz and others 2010; Barlow and McKenzie 2011; Koyasu and Moro 2011b). Thus, GATA-3 was identified as a critical transcription factor for their development (Hoyler and others 2012). Lin− c-kit+ Sca-1+ ILC2 cells were found in fat-associated lymphoid clusters in visceral adipose tissue. IL-33 activates ST2L on these cells, which induces the production of IL-5 and IL-6, but not IL-4, more than basophils or mast cells. This induction is independent of the combination of IL-2 and IL-25. However, IL-2 and IL-25 enhance the activity of IL-33 in this regard.
ILC2 is involved in defense mechanism against helminths via IL-5- and IL-13-dependent eosinophilia and goblet cell hyperplasia (Moro and others 2010; Koyasu and Moro 2011a). ST2-expressing ILC2 is present in mesenteric lymph nodes, spleen, and liver of IL-25 or IL-33 injected or helminth-infected mice. ILC2 is essential for host defense mechanism against Nippostrongylus brasiliensis infection (Neill and others 2010; Price and others 2010; Liang and others 2011) and the onset of allergic airway inflammation (Wilhelm and others 2011; Barlow and others 2012). However, there are ST2− Lin− c-kit+ Sca-1+ MPPtype2 cells present in the mesenteric lymph nodes and GALT of IL-25 or helminth-infected mice (Saenz and others 2010). It is believed that IL-33 and ST2 are vital factors for the expansion of IL-5- and IL-13-producing ILCs, but it is still controversial that ILCs express IL-5 and/or IL-13 under stimulation of IL-33 or IL-25.
Meanwhile, IL-33-responsive c-kit− Sca-1+ CD25+ cells were revealed as a distinct subset of cells from c-kit+ NH cells (Brickshawana and others 2011). In human and mouse lungs, Lin− c-kit+ Sca-1+ CD90+ CD25+ IL-7Rα+ ST2+ residential cells differentiated by a transcription factor, Id2, express IL-5 and IL-13 in response to IL-33 (Monticelli and others 2011). Two subsets of ILCs that solely express IL-5 or IL-13 in the presence of IL-33 or IL-25 in the peritoneal cavity, lungs, and gut of mice were also reported (Ikutani and others 2012). A subset of Lin− c-kit+ Sca-1+ CD25+ ILCs that expresses IL-5 and IL-13 in response to IL-33, but not to IL-25, from mouse lungs was also reported (Bartemes and others 2012) (Fig. 2).
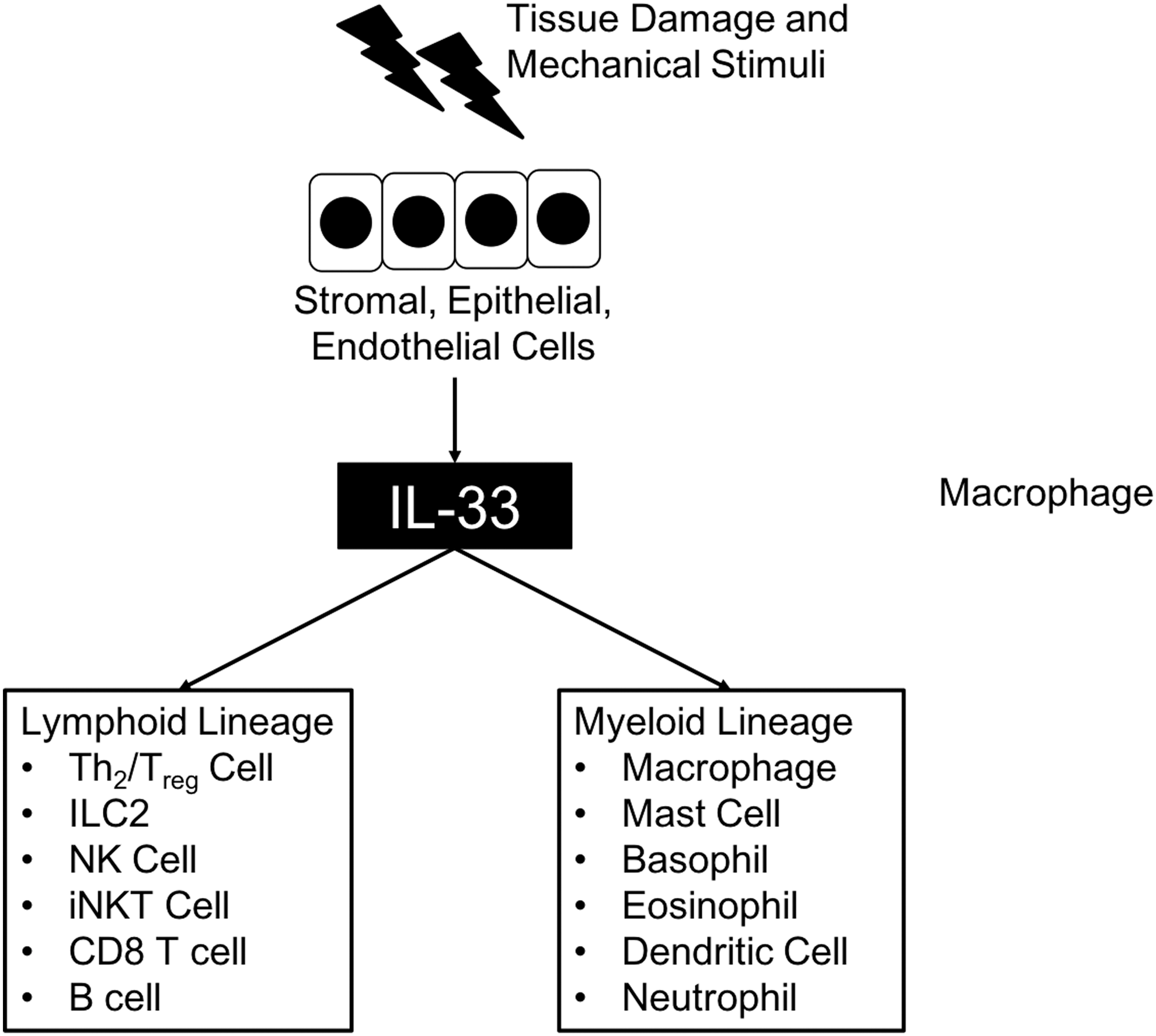
Cellular targets of IL-33. Stromal, epithelial, and endothelial cells secrete IL-33 after tissue damage or mechanical stress. Secreted IL-33 stimulates several subsets of both lymphoid and myeloid lineages of immune cells.
IL-33 and Cancers
Cytokines play central roles in cellular interaction in the inflammatory tumor microenvironment (Candido and Hagemann 2013). In recent reports, IL-33 is highly involved in several cancers where this cytokine exerts protumorigenic and antitumorigenic functions up to the environment (Lu and others 2016).
Breast cancer
Breast cancer is a primary cause of women's death by malignant tumors (Ferlay and others 2010). The reason for fatalness from breast cancer is extensive metastasis to the bones, brain, liver, and lungs (Lu and Kang 2007). The paradoxical roles of the immune system lead to the promotion or prevention of tumorigenesis. Leukocytes infiltrate in tumor tissues and have a substantial consequence on cancer development. CTLs and NK cells have antitumorigenic functions; meanwhile, Tregs and myeloid-derived suppressor cells have immunosuppressive functions in the tumor microenvironment (Jiang and Shapiro 2014; Carrega and others 2016).
IL-33/ST2 signaling has been reported to promote breast cancer in several studies. IL-33 and ST2 levels are increased in breast tissues with cancer than in healthy breast tissue (Liu and others 2014a; Kim and others 2015). Also, IL-33 and its decoy receptor ST2 levels are increased in breast cancer patients and correlated with the markers of poor prognosis, such as VEGF, MMP-11, and platelet-derived growth factor-C (Yang and others 2015).
When ST2 is deficient, it leads to delayed tumorigenesis and metastasis through downregulated tumor cell proliferation in a syngeneic 4T1 breast cancer model. Besides, the administration of IL-33 increased tumor cell proliferation in wild-type mice in this model (Jovanovic and others 2011, 2014). There is also a report that IL-33 has a straight effect on malignant breast cancer cells. Direct stimulation of ST2-positive breast cancer cells with IL-33 enhanced the size, proliferation, and colony formation. This happened via phosphorylation and activation of p38 MAPK pathway through IL-33/ST2 signaling (Kim and others 2015).
These findings show that the IL-33/ST2 pathway enhances breast cancer progression directly and indirectly. This noticeable change suggests that IL-33/ST2 can be a strong candidate for diagnosis and therapy of breast cancer.
Colorectal cancer
Colorectal cancer is one of the most common cancers and in many cases has a fatal prognosis (Brenner and others 2014; Wasmer and Krebs 2016). Colorectal cancer shows the classic developmental stages of epithelial cancer progression, adenoma-carcinoma consequence, followed by the accumulation of mutations in critical tumor genes and tumor suppressor genes such as Wnt/β-catenin, TP53, MAPK, KRAS, myc, and TGF-β/bone morphogenetic protein signaling pathways (Vogelstein and others 1988; Terzic and others 2010; Fearon 2011). Chronic inflammation like inflammatory bowel disease is a crucial factor of colorectal cancer (Grivennikov and others 2010; Grivennikov and Cominelli 2016; Robles and others 2016; Wasmer and Krebs 2016). Furthermore, several proinflammatory cytokines and inflammatory mediators are involved in colorectal cancer progression (Mager and others 2016; Wasmer and Krebs 2016).
Unlike other studies that show protumorigenic functions of the IL-33/ST2 pathway, there is a recent report that suggests a protective role of IL-33. IL-33 and ST2 levels are upregulated in adenoma and low-grade adenocarcinoma of patients with colorectal cancer (Cui and others 2015; Mertz and others 2016). Also, IL-33 messenger RNA (mRNA) level was upregulated in patients with stage I–III colorectal cancer, showing protective features (Zhang and others 2017). The high expression of IL-33 and ST2 is also reported in mouse colitis-related models. ST2-deficient mice showed delayed tumor growth and fewer lesions of colorectal cancer, whereas IL-33-overexpressed animals showed enhanced tumorigenesis and metastasis (Maywald and others 2015; Mertz and others 2016; Zhang and others 2017).
Metastatic SW620 human colorectal cancer cell line shows increased invasive potential not only with the stimulation of exogenous IL-33 but also with overexpression of IL-33. Furthermore, the overexpression of IL-33 in SW620 cells increased tumor growth, metastasis, and reduced survival when they were injected into nude mice (Liu and others 2014b).
Interestingly, in a recent finding, the expression of ST2 in tumor tissues was lower than the adjacent nontumor sites, which accompanied with a slowed tumor progression. This is because of reduced CCL2 production by tumor cells due to downregulated IL-33/ST2 signal (O'Donnell and others 2016). Also, IL-33-mediated IgA production leads to the prevention of microbial dysbiosis and IL-1α-dependent inflammation in the intestine. In AOM/DSS-treated IL-33-deficient mice, the secretion of inflammatory cytokine was increased, and tumor number, size, and grade were also increased compared with wild-type mice (Malik and others 2016). However, this does not necessarily mean that IL-33 has a similar function in humans. In mouse, manipulation of microbiota alone is sufficient to impair colorectal cancer progression, regardless of IL-33 (Mager and others 2016).
From these findings, it is not easy to determine if IL-33/ST2 signaling promotes or protects from colorectal cancer. It will be necessary to compare each mouse models' characteristics and their mechanisms that show protumorigenic or protective functions. Especially in human patients, chemotherapy may regulate the expression of ST2 and IL-33 levels, which likely influence tumor progression (O'Donnell and others 2016). More investigations are needed to understand the role and mechanism of IL-33/ST2 signaling in tumorigenesis to target this signaling pathway for efficient cancer therapy.
Non-small-cell lung cancer
Non-small-cell lung cancer (NSCLC) is the leading cause of lung cancer, 85% of lung cancers. The prognosis of NSCLC has not changed dramatically, but the underlying mechanisms have been understood prominently (Chen and others 2014). IL-33/ST2 in respiratory diseases was mainly studied regarding allergy, asthma, and chronic obstructive pulmonary diseases (Liew and others 2010, 2016; Byers and others 2013; Makrinioti and others 2014), whereas it has been studied much less in cancer.
Patients with NSCLC have enhanced expression of IL-33 and ST2 in cancer tissue than in adjacent healthy tissue, and the expression level is correlated with the clinical stage (Wang and others 2016). In opposition, another study showed decreased levels of IL-33 and ST2 mRNA in lung cancer tissue compared with healthy tissue, and the transcribed mRNA levels were reverse-correlated with cancer stages and survival rate. Along the same line, the protein level of ST2 was increased in low metastatic cancer cells than in highly metastatic 3LL Lewis lung carcinoma cells (Akimoto and others 2016).
The potential function of IL-33/ST2 in lung cancer remains controversial. Different patient cohorts, and the size of the cohort, might contribute to the discrepancy. There could be other possible variations like radiotherapy or surgery of the tumor tissue. Chemotherapy and radiotherapy can distort the molecular panel quite rapidly, which may also lead to the change of IL-33 level (Ha and others 2014; Tan and others 2016). Therefore, additional studies regarding IL-33/ST2 in lung cancer with primary NSCLC cells are needed to clarify its role and precise mechanism.
Other cancers
In hepatocellular carcinoma (HCC), many patients have a history of chronic hepatitis B or C infections followed by the progression of fibrosis or cirrhosis with high mortality. The multistep process of HCC involves the role of angiogenesis (Raza and Sood 2014; Waller and others 2015). Compared with healthy liver tissue, IL-33 expression was positive in the liver tissue of patients with HCC. Besides, serum IL-33 levels were higher in both preoperative and postoperative serum samples of HCC patients compared with healthy controls (Zhang and others 2012). The mechanism of IL-33/ST2 signaling in HCC is still unclear. However, in HCC tissues, effector-memory CD8+ T cells were increased, which may be associated with prolonged patient survival (Brunner and others 2015).
Among primary hepatic malignancies, cholangiocarcinoma (CCA) is the second most common that affects biliary duct system (Tyson and El-Serag 2011). CCA is closely associated with chronic inflammation, parasite infestation, primary sclerosing cholangitis, hepatolithiasis, biliary duct cysts, and poisonings (Tyson and El-Serag 2011). In mouse models, the ST2 levels increased in CCA, which enhanced IL-33 signaling and resulted in the elevation of IL-6 expression, as well as enhanced mitotic signals and cancer cell survival (Kobayashi and others 2005; Wehbe and others 2006; Yamada and others 2015). Also, IL-33-administered mice showed enhanced proliferation of cholangiocytes through an increased number of type-2 ILCs that produce IL-13. Activation of these ILCs activates AKT and YAP constitutively in bile ducts to promote liver metastasis (Li and others 2014). Moreover, external administration of IL-33 encouraged CCA development in mouse CCA models (Yamada and others 2015).
Head and neck squamous cell cancer (HNSCC) has high mortality and difficulties for surgical resection because of the anatomical complicity. The etiology of HNSCC includes smoking, alcohol, and human papillomavirus (Leemans and others 2011). Cancer-associated fibroblasts (CAFs) are critical participants in the HNSCC microenvironment promoting tumor growth, invasion, and metastasis, which means that CAF is a decisive factor in the severity of the disease. CAFs release IL-33 that induces epithelial-to-mesenchymal transition of cancer cells and cell aggressiveness. Besides, IL-33 induces its own expression and forms positive feedback. The expression level of IL-33 in CAFs is reversely correlated with the survival of patients. A poor prognosis is primarily indicated for patients with HNSCC in tongues with high expression of IL-33 and enhanced microvessel formation in the stroma (Chen and others 2013; Ishikawa and others 2014; Wasmer and Krebs 2016).
Gastric cancer is the fourth most common cancer with the second highest cause of death due to cancer throughout the world. The primary etiology factors of gastric cancer are diet, Helicobacter pylori infection, and gastroesophageal reflux and obesity (Carcas 2014). Meanwhile, inflammation is an essential factor in gastric cancer formation (Peek and Crabtree 2006; Fox and Wang 2007).
Renal cell carcinoma is the ninth most common cancer in men. In a recent report, IL-33 expression is tightly correlated with the poor prognosis for patients with renal cell carcinoma. The tumor cells have enhanced signaling of JNK, which has been stimulated by IL-33/ST2 signaling pathway (Wu and others 2018).
Conclusion
Many studies have investigated the role of IL-33, which provides novel insights into the mechanism of cancers. IL-33 is released from various types of cells during inflammation-mediated tissue injury. IL-33 signaling promotes the severity of allergic responses and cancer progression since IL-33 is an important factor to increase allergy-mediated inflammation that can worsen many types of cancer diseases.
The tumor microenvironment is a central operating system for the progression of tumor development, local invasion, and metastasis (Hanahan and Coussens 2012). One of the main regulators of the tumor microenvironment is pro- or anti-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α (Candido and Hagemann 2013). As an alarmin, IL-33 amplifies innate immune responses that can contribute to several different types of inflammatory disorders as well as modulation of tumorigenesis (Oboki and others 2010; Liew and others 2016; Lu and others 2016). Numerous studies using patient samples, in vitro experiments, and in vivo mouse models reveal multiple roles of the IL-33-ST2 pathway in the tumor microenvironment regarding tumor initiation, development, and resistance to therapy. In general, IL-33 has a protumorigenic feature in different types of cancers; however, contradictory findings of IL-33 were also reported in several other types of cancers (Wasmer and Krebs 2016). It is not easy to develop a new therapy before all research processes, including animal experiments and clinical trials, have been explored. However, patient- or disease-specific therapy using IL-33 or the antagonist (ST2) can be an additive solution for cancer treatment.
The mechanism of IL-33 in cancer is still indistinct. It is always controversial in that IL-33 maturation is obtained by particular enzymatic activity. Nevertheless, this cytokine may be activated by multiple activating processes. In addition, the various effects of IL-33 in T cell immunity suggest the possible existence of another component of IL-33R complexes. Unlike the identified function of ST2 and IL-1RAcP complex, another member of the IL-33R complex may be the clue to explaining the effect of IL-33 on both Th1 and Th2 immunity regarding inflammation-mediated cancers.
Footnotes
Acknowledgments
J.H. and P.C.L. are supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. S.K. was supported by the National Research Foundation of Korea Grants NRF-2015R1A2A2A01003472, -2014M3A6A4075058, and -2015R1A2A1A15051472.
Author Disclosure Statement
No competing financial interests exist.