
Abstract
Hepatitis C virus (HCV) is a global health problem, with an estimated bioburden of >180 million. Every year about 350,000 people die from HCV-associated liver complications such as cirrhosis and cancer (e.g., hepatocellular carcinoma). Pakistan has the second highest prevalence of HCV. Treatment of this life-threatening disease has been a challenge, but recent developments in direct-acting antivirals have offered hope to many. Although direct-acting antivirals have dramatically improved viral clearance, their exorbitant costs put them out of reach for patients in developing countries. Thus, interferon therapy is still being used in Pakistan. Specifically, interferon-stimulating genes can alter treatment response. For example, interferons induce expression of many antiviral genes through signal transducer and activator of transcription/Janus kinase signaling. Suppressor of cytokine signaling genes play an eminent role in the inhibition of cytokine signaling pathways and regulation of both adaptive and innate immunity. The present review examines expression of suppressor of cytokine signaling-1 in HCV-treated patients.
Hepatitis C virus (HCV) is an alarming health problem, with a global bioburden of >180 million people, representing >3% of the world's population. HCV infection can be mild to serious, causing cirrhosis, hepatocellular carcinoma (HCC), and other liver disorders. More than 350,000 people succumb to HCV-associated liver complications annually, and only a small number of patients are able to clear HCV spontaneously during acute infection. While release of cytokines, especially interferons (IFNs), acts as an innate immune response that clears HCV during acute infection, many patients fail to eliminate the virus completely. Thus, HCV persists in 50%–80% of infected people and causes chronic infections. Of the chronic HCV (cHCV) patients, 4%–20% develop cirrhosis within 10–20 years, and 1%–5% also develop HCC. Viral factors and strategies behind HCV persistence include interruption of IFN-stimulated gene production, induction of IFN signaling pathways, and direct antagonism of effector systems by viral proteins. To date, an anti-HCV vaccine has not yet been developed. However, therapeutic options are constantly improving. Although IFN therapy has been the standard of care for cHCV patients, this treatment has a high relapse rate and significantly low rate of sustained virologic response (SVR). These setbacks have led to the development of IFN-free regimens, specifically direct-acting antivirals (DAA), which have revolutionized HCV therapeutics.
Host metabolic factors, various single nucleotide polymorphisms, and gene expression patterns can help predict treatment outcomes in HCV patients. In particular, suppressor of cytokine signaling (SOCS) proteins play an important role in the maintenance of organ homeostasis by acting as negative regulators for various cytokine responses, and SOCS genes play an eminent role in the inhibition of cytokine signaling pathways and regulation of both adaptive and innate immunity. For example, these molecules are involved in dendritic cell activation, positive and negative regulation of macrophages, and T-cell development and differentiation. Several studies have suggested involvement of SOCS proteins in immunodeficiency and autoimmune disorders. Cytokines play an important role in lymphoid and myeloid cell function, differentiation, and development, and hematopoietic growth factors, IFNs, and interleukins (ILs) are cytokines that activate Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathways. Cytokines trigger large numbers of SOCS genes and inhibit cytokine signal transduction through classical negative feedback loops. Other stimuli that trigger activation of SOCS proteins are cyclic AMP, isoproterenol, statins, and lipopolysaccharides. SOCS proteins are an integral part of many biological processes. For example, in the absence of cytokine signaling, SOCS1 expression is developmentally regulated in immature thymocytes. SOCS1 negatively regulates Toll-like receptor (TLR)-mediated signaling and cytokine receptors, and several cytokines (e.g., IFN-γ and IL-4) and TLR ligands, such as CpG-DNA and lipopolysaccharides, induce expression of SOCS1.
Some studies have shown an inverse relationship between SVR rate and expression of SOCS1 and SOCS3 in the livers of cHCV patients. However, literature discussing sequential changes in the expression of SOCS genes before and after treatment with IFNs and ribavirin in cHCV patients is lacking. The first study to demonstrate SOCS1 expression in HCV patients resistant to IFNs was published in 2010 (Fujimoto and Naka 2010), and Table 1 summarizes the role of SOCS1 expression in the pathophysiology of the liver in HCV-infected patients.
Function of SOCS1 in the Pathophysiology of the Liver in Hepatitis C Patients
SOCS, suppressor of cytokine signaling; TLR, Toll-like receptor.
Imanaka and others (2005) revealed that SOCS family molecules act as important elements in JAK–STAT signaling, specifically IFN signaling, and reported negative expression of SOCS genes in cHCV patients treated with IFN-α. Specifically, expression levels of SOCS1 and SOCS3 were investigated in the livers of 21 cHCV patients and 8 healthy controls by reverse transcriptase polymerase chain reaction, as well as tyrosine phosphorylation of IFN-α-induced STAT1 in human hepatoma cells (PLC/PRF/5) stably transfected with SOCS1 via immunoblotting. In addition, Northern blot was used to study the expression of antiviral genes. Analysis of the clinical outcomes of 77 cHCV patients treated with IFNs showed significantly higher SOCS1 expression in cHCV patient livers compared to healthy controls (P < 0.005). Moreover, the SVR rate was significantly lower in cHCV patients with high SOCS1 levels versus patients with low SOCS1 expression (P = 0.0014). Thus, upregulation or increased expression of SOCS1 in the liver may be related to IFN resistance in cHCV patients. Furthermore, Tseng and others (2013) demonstrated that heightened methylation of the SOCS1 promoter was directly related to SVR at the end of IFN therapy in 106 cHCV patients.
Yao and others (2005) first presented a novel mechanism of HCV persistence wherein ligation of HCV core protein to complement receptor gC1qR on monocytes/macrophages induces SOCS and results in inhibition of T-cell function. Zhang and others (2015) further revealed that persistent HCV infection results from dysregulation of innate immune responses. Expression of IL-12 is impaired by HCV core protein interaction with gC1qR. Moreover, negative immunomodulators SOCS1 and programmed death-1 (PD-1) are involved in dysregulation of lymphocytes mediated by the HCV core. Zhang and others (2015) investigated the role of SOCS1 and PD-1 in IL-12 suppression mediated by the interaction between HCV core protein and monocytes/macrophages, and also estimated the THP-1 cell line or TLR-stimulated, primary CD14+ monocytes/macrophages from healthy and cHCV patients following HCV core treatment. They revealed that increased baseline PD-1 expression in monocytes/macrophages of cHCV patients was significantly correlated with the degree of IL-12 inhibition. In particular, increased PD-1 and SOCS1 expression concomitant with decreased IL-12 expression was found in monocytes/macrophages from healthy and HCV-infected people treated with HCV core protein. The same results were found in cells treated with gC1qR ligand-C1q. Blocking of PD-1 signaling decreased SOC1 expression and enhanced IL-12 production induced by HCV core protein, whereas blocking of gC1qR suppressed IL-12 and rescued HCV core protein-induced PD-1 upregulation. In contrast, increased expression of IL-12 and inhibition of PD-1 upregulation occurred through silencing of SOCS1 expression with the use of small interfering RNAs. Furthermore, activation of STAT1 during TLR-stimulated IL-12 production occurred in response to silencing of SOCS1 and blockade of PD-1 in monocytes/macrophages.
Xu and others (2014) demonstrated upregulation of microRNA (miR)-221 in cHCV patients and Huh cell lines infected with HCVcc. In particular, SOCS1 and SOCS3 were found to be targets of miR-221 that resulted in accentuation of the anti-HCV effect of IFN therapy. In addition, Ren and others (2016) found upregulation of miR-146a in monocytes of HCV patients versus healthy controls. SOCS1 and miR146a check excessive activation of STAT3 and trigger appropriate immune responses in vivo. The study demonstrated that miR146a may regulate the SOCS1/STAT3 signaling and the expression of cytokine in monocytes resulting in differentiation of T cells and balancing of immune injury and immune clearance during viral infections. The study also showed that miR146a inhibition in monocytes of CHC patients was associated with decreased expression of IL-23, TGF-β, and IL-10 through the inhibition of signal transducer and activator transcription 3.
Iijima and others (2015) analyzed early treatment outcomes related to IFN-stimulated genes and IFN-λ in peripheral blood mononuclear cells (PBMCs) of cHCV patients who received IFN therapy with ribavirin and NS3/4 protease inhibitor. About a quarter of patients with a favorable genotype (TT or TG/GG at rs8099917) and an unfavorable IL28B genotype (TG, GG, or TG/GG at rs8099917) achieved SVR. IFN regulatory factor 1 expression was found to be significantly higher in patients with an unfavorable IL-28B genotype than those with a favorable one. Moreover, they reported that SOCS1, SOCS3, and A20 mRNA levels interfered with IFN signaling pathways and suppressed antiviral activity, especially in non-SVR or non-responders. These mRNA levels also varied according to IL-28B genotype during pegylated-IFN/ribavirin/protease inhibitor therapy and effected treatment outcomes. Lee and others (2015) reported increased SOCS1 expression in SVR versus non-SVR patients who received 24- to 28-week pegylated-IFN/ribavirin treatment after infection with HCV genotype 1. Lee and others (2015) evaluated the expression of JAK-STAT signaling genes in PBMCs and revealed nonsignificant difference between SOCS1 expression in SVR and non-SVR patients. The findings of this study contradict the previously reported findings that showed increased rate of SVR in HCV patients who express SOCS1 gene.
Georgel and others (2010) reviewed different cellular and viral factors that play an important role in host–virus interactions and uncovered a mechanism used by HCV to cause infection and escape both adaptive and innate host immunity (Figs. 1 and 2).

HCV evasion factors target the host innate immune response: two important pathways of innate immunity involved in sensing HCV infection. Signals are transmitted through adaptor molecules (TIR domain-containing adapter-inducing interferon (IFN)-β and mitochondrial antiviral-signaling protein/IFN-β promoter stimulator 1/virus-induced signaling adapter) after Toll-like receptors and/or RIG-1-like receptors detect virus-derived epitopes. Signals are cleaved by NS3/4A protease, which impedes early type I (a/b) and type III (l) IFN synthesis, whose production is controlled by common signaling pathways (Lee and others 2016).
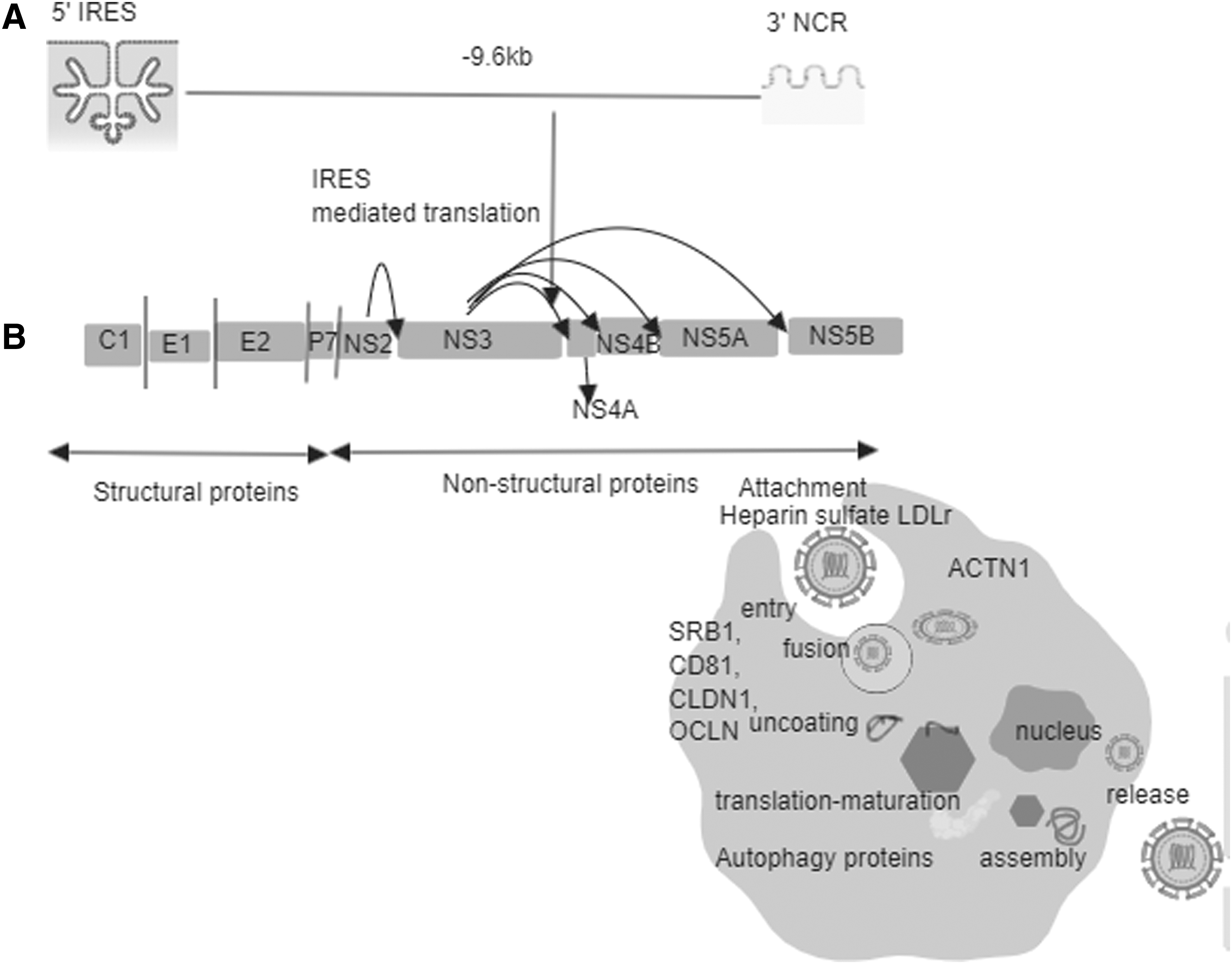
HCV genome and polyprotein.
Li and others (2013) reported that SOCS1 is a target of liver-specific miR-122. The efficacy of IFN therapy is known to be poor in cHCV patients with low miR-122 levels. Their study observed significantly heightened IFN expression in the miR-122 rich Huh-7 cell line, whereas decreased IFN expression was found when miR-122 was knocked down. Thus, miR-122 is thought to modulate SOCS1 expression, which regulates subsequent IFN-1 expression.
Yoshikawa and others (2001) indicated transcriptional silencing was a result of aberrant methylation in the CpG island of SOCS1. Their study observed aberrant methylation expression in 65% of 26 human primary HCC tumor samples. Restoration of SOCS1 expression suppressed growth rate and anchorage-independent growth of cells in those tumor samples having methylation-silenced SOCS1 expression or constitutively activated JAK2 gene. Suppression of growth occurred as a result of apoptosis and was reproduced by administration of the JAK2 inhibitor AG490, which reversed the constitutive phosphorylation of STAT3 in SOCS1-inactivated cells. This study suggested a therapeutic strategy involving SOCS1 in gene therapy and inhibition of JAK2 by small molecules (e.g., AG490 for treatment of HCC).
Shao and others (2013) found that overexpression of SOCS1 had no effect on HCV RNA replication but did rescue suppression of HCV replication by IFN-α. In addition, Nagai and others (2002) reported silencing of STAT-induced STAT inhibitor-1 (also called SOCS1) in a significant portion of HCC samples (50%) through combined mechanisms comprised of chromosomal loss of the remaining allele of the SSI-1 gene and methylation of either 50 or exon CpG-rich regions. Miyoshi and others (2005) showed the ability of HCV core protein to downregulate SOCS1 expression that may lead to its silencing, which is an alternative to SOCS1 silencing through hypermethylation. Their study further clarified the effect of HCV core protein modulation of intracellular signaling pathways and pathogenesis of HCV infection, including hepatocarcinogenesis. Finally, Formeister and others (2010) tested gene expression and promoter methylation endpoints in 43 tumorous and 45 non-tumorous liver tissue samples from HCC patients with HCV. In addition to tumor recurrence, the promoter methylation status of many genes, including SOCS1, was monitored. Their results showed that SOCS1 and other gene were more frequently methylated in non-tumorous tissue.
Researchers from Tokyo studied the relationship between IFN-λ4 and treatment outcome and its effect on IFN-stimulated gene expression in liver samples collected from 49 cHCV patients treated with IFNs (Murakawa and others 2017). SNPs associated with interferon lambda 4 (IFNL4) gene act as predictors of treatment outcome with both interferon-based regimen and interferon-free regimens. IFNL4 gene encodes IFNλ4 and rs368234815-TT/ΔG control its expression. The study analyzed the association and effects of IFNL4 gene on intrahepatic interferon stimulated gene (ISG) expression and demonstrated its correlation with treatment outcome in patients treated with Peg-IFN/ribavirin. The expression of IFNL4 was associated with treatment failure towards Peg-IFN/ribavirin. The findings of the study suggested that decreased ISG expression was associated with treatment failure whereas, intrahepatic expression of IFNL4 increased antiviral ISG expression.
Likewise, another group of researchers quantified the transcriptional levels SOCS1 and IFN-α receptor 1 (IFNAR1) in the PBMCs of treated and untreated HCV patients and healthy controls (Sedeño-Monge and others 2010). Treated HCV patients were categorized as responders or non-responders based on the viral load measured 6 months after IFN treatment completion. Their results showed that transcriptional levels of IFNAR1 were higher in responders, non-responders, and untreated HCV patients versus healthy individuals. In addition, patients infected with HCV genotypes 1 and 1b had significantly higher (P = 0.003) IFNAR1 transcription compared to patients infected with HCV genotype 1a1b. These findings suggest that IFNAR1 and SOCS1 transcription, as well as the ability to evade antiviral response mechanisms, differs by HCV genotype. Massirer and others (2004) examined IFNAR1 mRNA expression in PBMCs of cHCV patients treated with IFN therapy and showed that responders had significantly higher IFNAR1 mRNA expression than controls and non-responders before treatment. Another study by Morita and others (1998) in IFN-treated patients infected with HCV genotype 2 showed significantly higher IFNAR1 and IFNAR2 expression (P < 0.01) in the liver of responders versus non-responders. Importantly, heightened expression of these genes was a predictor of complete response to IFN treatment, with a positive predictive value of 100%. Furthermore, Mizukoshi and others (1998) reported increased IFN-α/β receptor mRNA expression in sustained versus unsustained responders (P < 0.01). Thus, IFN-α/β receptor mRNA expression may also act as an important host factor that determines response to IFN therapy.
Pfeffer and others (2014) found dysregulated expression of a subset of IFN-responsiveness genes in liver samples of IFN treatment responders and non-responders. Importantly, they developed a statistical model based on gene expression data compared to an IL-28 mutation model that helps predict response to IFN therapy. Lower expression of STAT1 and XAF1 is associated with IFN-responsiveness towards IFN-therapy whereas, increased expression of RSAD2, IFI6, IFI16, and CCL5 was associated with poor response towards therapy.
Recently, a study was published showing increased SOCS1 expression with an increase in HCV load (Wahid and others 2018). Another study demonstrated 5.4- and 1.2-fold higher SOCS1 expression in IFN- and DAA drug-treated patients, respectively (Naz and others 2018). This study also revealed a significant difference in the relative expression of SOCS1 and SOCS3 in DAA drug- and IFN/ribavirin-treated and untreated individuals. These results indicate that expression of SOCS1 and SOCS3 can be normalized by targeting HCV proteins with DAA drugs compared to IFN/ribavirin therapy.
Conclusion
SOCS proteins negatively regulate inflammatory signaling pathways by facilitating proteasomal degradation and ubiquitination of pathway machinery. HCV evades host immune response because of its ability to mediate inhibition of JAK/STAT pathway. Existing literature suggest that SOCS1 and SOCS3 genes possess considerable therapeutic potential. Analysis of SOCS genes expression may act as predictor of treatment response in HCV infected patients. Increasing the expression of SOCS gene during viral infection improves treatment response in HCV patients however, more research is needed to further interrogate the mechanism behind association of SOCS gene expression with DAA drugs. In particular, overexpression of SOCS1 is found in PBMCs of HCV patients that do not respond to IFN therapy and positively correlates with viral load.
Footnotes
Author Disclosure Statement
No competing financial interests exist.