
Abstract
Interleukin-12 (IL-12) is a pleiotropic cytokine that has profound effects on many aspects of cell-mediated responses and can enhance antitumor responses in experimental models. IL-12 has been tested clinically, however, side-effects have limited its use. We are developing an attenuated form of IL-12 whose biological activity could be restricted to sites of tumors by taking advantage of overexpressed tumor proteases that can activate the cytokine. We constructed a panel of fusion proteins (FPs) consisting of IL-12 joined to a specific inhibitor connected by a protease cleavage sequence (cs). We first identified a panel of single-chain Fragment variable (scFv) that bind to 3 independent epitopes on IL-12 and then incorporated them into separate IL-12 FPs containing either a matrix metalloproteinase (MMP) cs or a scrambled (scram) control cs. The intact IL-12 FPs showed attenuation in IL-12 activity compared to free IL-12 in 2 separate in vitro functional assays; proliferation of CTLL-2 and interferon-gamma (IFN-γ) induction by spleen cells. Furthermore, the FP containing the MMPcs showed an increase in biological activity of IL-12 in vitro when cleaved by MMP9. This FP strategy could be applied to other immunomodulators and potentially reduce unwanted side-effects observed with systemic delivery thus improving cytokine immunotherapy strategies.
Introduction
Cytokines have long been known to be key immune mediators and are involved in almost all aspects of immune responses. As a result, many have been tested in a variety of cancers with some notable successes. For example, interleukin-2 (IL-2) is now FDA approved for the treatment of melanoma and kidney cancer (Rosenberg and others 1994; Rosenberg 2001). However, side effects have greatly limited their utility and efficacy and are a fundamental concern in using cytokines systemically (Baluna and Vitetta 1997; Panelli and others 2004; Skrombolas and Frelinger 2014). These systemic effects are not totally unexpected given that most cytokines have evolved to act locally over short distances in an autocrine or paracrine fashion (Pardoll 1995).
Strikingly, local expression of cytokines in the tumor microenvironment, achieved by a variety of means, can be remarkably effective at generating antitumor immune responses and even result in tumor rejection of the treated tumor and the generation of memory responses (Gansbacher and others 1990; Egilmez and others 2000; Hanes and others 2001; Moran and others 2003; Simpson-Abelson and others 2009; Gerber and others 2013).
Nevertheless, because cancerous lesions are often numerous and most may not be directly accessible, translating these advances in cytokine therapy has proven challenging. Initial attempts to extend this cytokine approach have been based on the rationale that treating a few accessible tumor lesions would engender a vigorous systemic immune response that in turn would eliminate even untreated tumor sites. Unfortunately, however, the general finding is that the treated tumor site is almost always more affected than the unmanipulated tumor sites. Thus, there is a critical need to develop ways in which the cytokine milieu of all tumor sites can be altered using a systemic approach.
A conceptually appealing strategy has been to couple cytokines to antibodies that bind tumor-associated antigens (termed immunocytokines) with the goal of locally increasing cytokine levels at the tumor site (Adams and others 2003; Dela Cruz and others 2004; Schrama and others 2006; Hank and others 2009; List and Neri 2013). However, one underappreciated limitation of this approach is that the antibody does not “home” directly to the tumor target per se. Rather, the immunocytokine diffuses throughout the body and eventually accumulates at the tumor site due to the binding and retention of the antibody to its target. In this approach, the cytokine is of necessity always active (Skrombolas and Frelinger 2014; Tzeng and others 2015). As a result, it can bind to any cells in the blood or tissues with specific, high-affinity cytokine receptors, which may limit its effectiveness and also cause unwanted side effects.
We are developing a new and fundamentally different strategy that has the potential to increase cytokine levels at disseminated tumor sites (Puskas and others 2011; Skrombolas and Frelinger 2014). This novel approach employs a fusion protein (FP) in which a cytokine is joined to a specific inhibitory binding component separated by a protease cleavage site. We hypothesize that before cleavage, the cytokine would be largely inactive due to specific binding of the inhibitory component. However, after cleavage by proteases that are overexpressed at the tumor site, the cytokine could become available to interact with high-affinity receptors on immune cells.
Proteases are overexpressed in a variety of tumor types and can influence many critical processes involved in tumor progression, including invasion, proliferation, and angiogenesis (Koblinski and others 2000; Hojilla and others 2003; Mason and Joyce 2011). These proteases include cathepsins, matrix metalloproteinases (MMPs), caspases, and urokinases (Sloane and others 2005; He and others 2007; Li and Yuan 2008; Kessenbrock and others 2010; Mason and Joyce 2011). For example, proteases can act directly by degrading basement membrane components to enhance tumor spread or they can cleave cellular receptors resulting in their activation.
Proteases can also work indirectly by increasing the availability of growth factors by releasing them from the extracellular matrix or by initiating protease cascades whereby 1 protease converts downstream proteases into active forms, that in turn promote tumor growth or invasion (Koblinski and others 2000; Li and Yuan 2008; Mason and Joyce 2011). As a result of these direct and indirect effects, protease overexpression and dysregulation is often associated with tumor growth and aggressiveness and is an intrinsic property of many tumors.
We have previously developed a FP containing IL-2 that could become functionally more active after protease cleavage (Puskas and others 2011; Skrombolas and Frelinger 2014). However, a critical unanswered question is whether this approach could be generalized and used for other cytokines and thus serve as a platform technology. In this study, we set out to test this concept by employing IL-12 and a single-chain Fragment variable (scFv) as the inhibitory component.
IL-12 has potent effects on a variety of immune responses and is a key cytokine linking the innate and adaptive immune response (Vignali and Kuchroo 2012). IL-12 is one of the most robust polarizing cytokines enhancing T helper 1 (TH1) cells and also promoting development and activity of Cytotoxic T Lymphocytes (CTL) and Natural Killer (NK) cells (Mescher and others 2006). IL-12 can also enhance the number and diversity of responding T cells. As shown in the lymphocytic choriomeningitis virus (LCMV) system, IL-12 by itself, or in conjunction with other cytokines, can activate virus-reactive effector and memory cells to produce interferon-gamma (IFN-γ) independent of T cell receptor stimulation (Freeman and others 2015).
Particularly important for immunotherapy approaches, IL-12 has also been reported to reverse anergy in T cells, reactivate tumor-associated quiescent effector memory T cells, reprogram inhibitory cells such as tumor-associated macrophages (TAMs) from an immunosuppressive M2-like phenotype to a proimmunogenic M1-like phenotype and also alter the cytokine milieu of the tumor (Watkins and others 2007; Simpson-Abelson and others 2009; Vignali and Kuchroo 2012; Lampreht Tratar and others 2017; Jarosz-Biej and others 2018).
IL-12 has been tested clinically, but serious side effects limited its use (Cohen 1995; Leonard and others 1997) due to its potent pleiotropic effects and the cytokine storm engendered when delivered systemically. Strategies to limit these side effects are essential to advance the application of powerful immune mediators such as IL-12.
In the current study, we examined whether we could construct an IL-12 FP in which the cytokine was functionally inhibited in the intact form of the FP but could become more active after cleavage by a MMP. To achieve this goal, we first developed a functionally active fusion of the p40 and p35 subunits of IL-12 and isolated and epitope mapped a panel of scFv that react with IL-12. From this panel of scFv, we generated a series of IL-12/scFv FPs containing a specific MMP cleavage sequence or a scrambled control sequence (Bremer and others 2001). This panel of FPs was characterized and we demonstrated that a FP containing IL-12 could be specifically cleaved by MMP9 resulting in enhanced IL-12 biological activity.
Materials and Methods
Identification of scFv using phage display library
We used a phage library previously generated and characterized to identify scFv specific for IL-12 (Haidaris and others 2001; Shea and others 2005). Briefly, this antibody library was generated by polymerase chain reaction (PCR) amplification of the VL and VH regions of pooled leukocyte complementary DNA (cDNA) and these variable regions were connected by a 14 amino acid linker (EGKSSGSGSESKAS) between the VL and VH and cloned into the phagemid display vector AP-III6 (Haidaris and others 2001). Pooled transformants (∼2 × 109) were infected with helper phage (VCS M13) to produce the phage antibody library that was used for panning to select IL-12-specific scFvs. Phage enzyme-linked immunosorbent assay (ELISA) was used to identify scFv specific for either the p40 or p35 subunits of IL-12 as described previously with minor modifications (Puskas and others 2011).
Construction of mouse IL-12 FPs
The mouse single-chain FP connecting the p40 and the p35 subunits resulting in full-length IL-12 with a 6x Histidine (6x His) tag was constructed as described previously (Skrombolas and others 2015). The Gibson assembly strategy (Gibson and others 2009) was used to construct 6 different mouse IL-12 (mIL-12) FPs. In brief, PCR amplification using the mIL-12 forward and reverse primers (Table 1) generated the mIL-12 fragment. The scFv fragments (scFv1, scFv7, and scFv8) were amplified with individual forward primers and the same scFv reverse primer (Table 1).
Polymerase Chain Reaction Primers for Construction of Fusion Proteins
mIL-12, mouse interleukin-12; scFv, single-chain Fragment variable.
The amino acid sequences for scFv1, scFv7, and scFv8 are listed in Supplementary Fig. S1. The 6 gene blocks were purchased from Integrated DNA Technologies (IDT). Each Gibson reaction consisted of the mIL-12 fragment, the linearized pVL1392 vector and a combination of the scFv and gblocks for a total of 4 fragments per reaction.
Baculovirus production and purification of IL-12 and IL-12 FPs
The generation of recombinant baculoviruses for the expression of proteins in insect cells was performed as described previously (Rose and others 1990, 1994; Neutra and others 1992). We have previously used this system to produce biologically active single-chain IL-12 (Skrombolas and others 2015), so we used this approach to produce the IL-12/scFv FPs. Recombinant viruses were created using the pVL1392 transfer vector and the BD BaculoGold transfer vector system (BD Biosciences) basically as described by the manufacturer. Initial virus production was performed in Spodoptera frugiperda (Sf9) cells cultured in Sf-900 II serum-free media (SFM; Gibco). After 2 passages (P0, P1) of Sf9, a high-titer stock was obtained (P1).
For final protein production, Trichoplusia ni (Tni) cells were cultured in Express Five SFM (Gibco) plus 2 mM
Western blotting
Immunoblot analysis was performed as described previously with minor modifications (Puskas and others 2011; Skrombolas and others 2015). A monoclonal mouse anti penta-His primary antibody (Qiagen) was used for detection of the 6x His tag on the scFv portion of the FP followed by detection with a goat anti mouse horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch) or a monoclonal rat anti mIL-12 primary antibody (C17.8; BD Pharmingen) was used for detection of mIL-12 followed by detection with a goat anti-rat HRP-conjugated secondary antibody (Jackson ImmunoResearch) and both were developed using the Amersham ECL Prime Western blotting detection reagent (GE Healthcare) as per the manufacturer's instructions.
CTLL-2 proliferation assay
Functional IL-12 was measured using CTLL-2 cells (ATCC, Manassas, VA) as described previously with minor modifications (Khatri and others 2007; Skrombolas and others 2015). In brief, Ni-NTA-purified proteins or MMP cleaved products were serially diluted 1:5 in 50 μL of medium, then 4 × 104 CTLL-2 cells in 100 μL medium were added per well to a 96-well plate and incubated at 37°C in 5% CO2 for 18–22 h. Thiazolyl Blue Tetrazolium Bromide (MTT; Sigma-Aldrich) was added at 75 μg per well and the plate was incubated for 8 h at 37°C in 5% CO2. Cells were lysed with 100 μL per well of 10% sodium dodecyl sulfate (SDS; Gibco) acidified with HCl, incubated at 37°C in 5% CO2 overnight, and absorbance was read at 570 nm.
Cytokine production by spleen cell cultures
Equal molar amounts of either IL-12 or the IL-12 FPs were serially diluted in media in a 96-well plate. Spleen cells were added at 1 × 106 per well and the plate was then incubated at 37°C, 5% CO2 for 24 h. Supernatants were collected and an IFN-γ ELISA (eBioscience) was performed as per the manufacturer's recommendations.
In vitro digestion conditions for FPs
For digestion of the FPs containing the MMPcs1 cleavage sequence or the scrambled control sequence, MMP9 (R&D Systems) was activated with p-aminophenylmercuric acetate (APMA) and this activated protease or control activation buffer was added to the FPs and incubated in 50 mM Tris, 10 mM CaCl2, 150 mM NaCl, and 0.05% Brij35, pH 7.5 for 1 h at 37°C. The digests were then separated by SDS–polyacrylamide gel electrophoresis, transferred, and probed using antibodies to the 6x His tag or the cytokine as previously described.
Results
Identification and epitope mapping of scFv that bind IL-12 p70
A major objective of the current study is to determine the feasibility of constructing an IL-12 FP that is inhibited in the uncleaved state but could be activated by cleavage with an MMP. A schematic of the FP we are developing is shown in Fig. 1. As illustrated, it consists of an IL-12 heterodimer joined to a specific binding component forming a single molecule. We and others have shown that a protein fusion of the p40 and p35 subunits could be functionally active (Lieschke and others 1997; Skrombolas and others 2015). The next critical step in constructing an activatable IL-12 FP was to identify a component that would bind specifically to IL-12 and thus potentially block its function. For this purpose, we utilized phage display technology to identify scFv that bind IL-12.
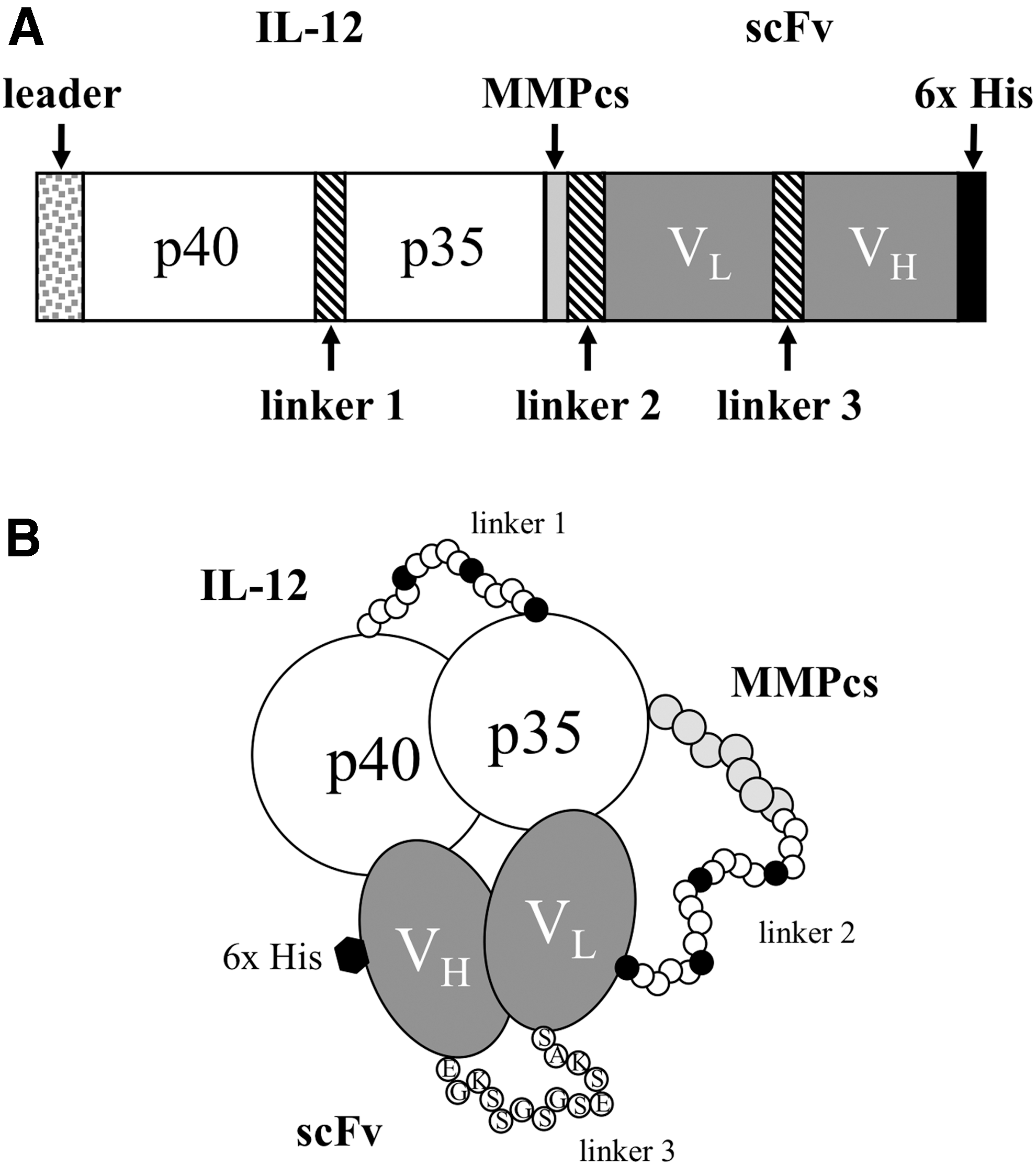
General structure of the IL-12 FP designed to be activated by MMPs.
An M13 phage display library was screened and enriched for binding to mIL-12 p70 by 2 rounds of panning. From this enrichment, a panel of 15 phage clones with unique sequences, indicating that they were independent isolates, were selected for further characterization. These isolated phage clones were assessed for binding to IL-12 by direct phage ELISA. In the direct phage ELISA, the antigen is first plated, followed by the addition of phage-displaying scFv (analogous to a primary antibody), and binding detected by subsequent addition of an enzyme-conjugated antibody to the M13 phage. As expected, since the phage library was panned on IL-12, all of the clones bound to IL-12 p70 (Fig. 2A).
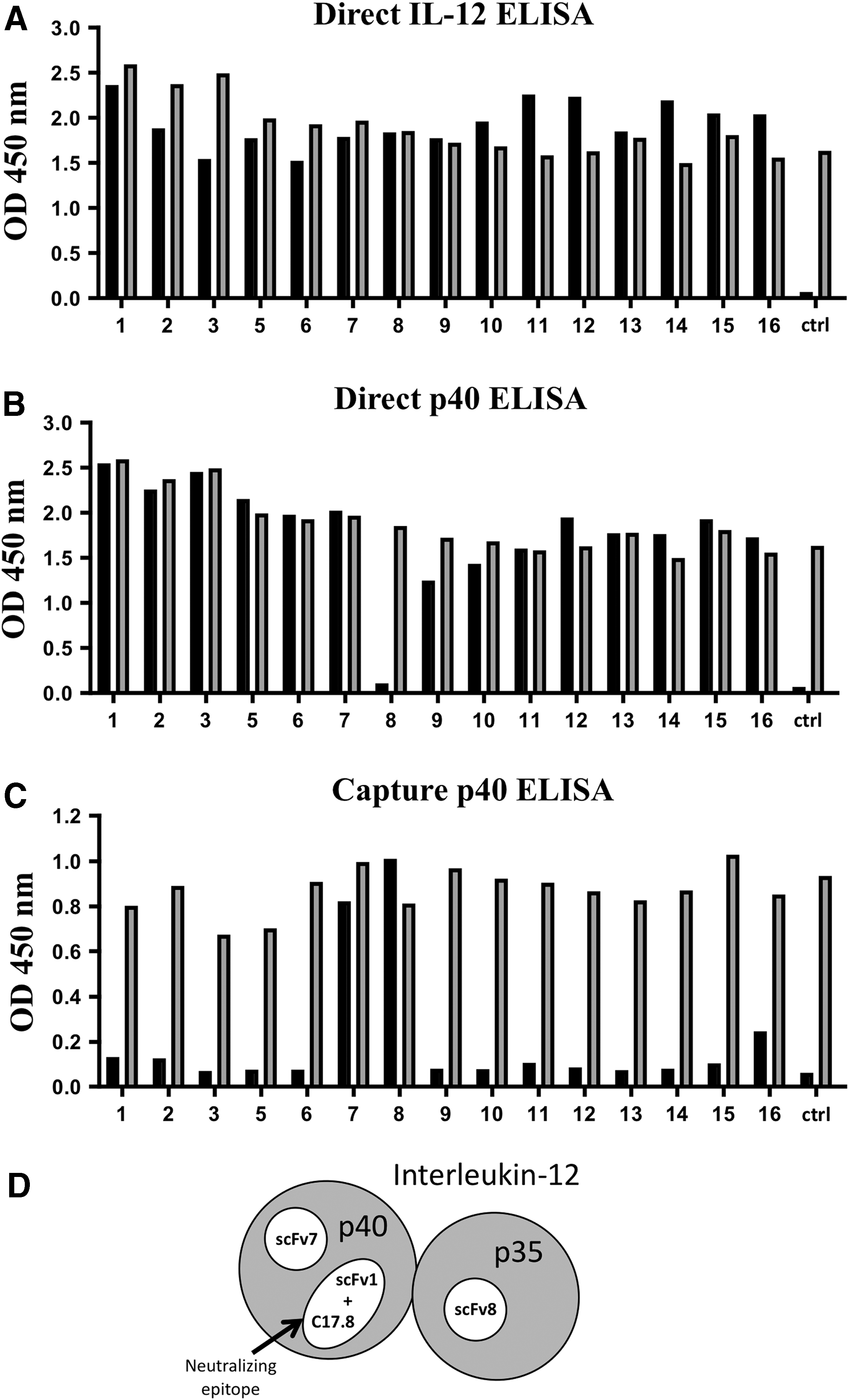
Epitope mapping of scFv reactive with IL-12.
To further examine their binding characteristics, we also tested these clones on p40 in the direct phage ELISA. Interestingly, all but one of the clones (clone 8) bound to the isolated p40 subunit, demonstrating that the majority of clones isolated recognize an epitope on p40 (Fig. 2B). However, clone 8 did not react with p40 in the direct ELISA suggesting it may bind to an epitope on p35.
We further analyzed these clones using a capture IL-12 ELISA employing a neutralizing monoclonal antibody (anti mIL-12 [C17.8]) that reacts with the p40 subunit as the capture antibody (Fig. 2C). Strikingly, almost all of the clones tested were negative in this assay. These results indicate that most of the scFv react with a similar, if not an identical epitope, as the anti-p40 monoclonal antibody (C17.8).
However, clone 7 and clone 8 did react in this capture ELISA (Fig. 2C). Since clone 7 reacts with isolated p40 (Fig. 2B), these results indicate that clone 7 likely reacts with a second epitope on p40 distinct from the one recognized by C17.8. In contrast, clone 8 did not react with p40 in the direct ELISA; however, it did react in the IL-12 p70 capture ELISA consistent with the idea that it recognizes an epitope on p35. Based on these data, we chose clone 1 (p40 binder, likely recognizing a neutralizing epitope), clone 7 (a second independent p40 binder), and clone 8 (a putative p35 binder), as scFv to incorporate into IL-12 FPs (Fig. 2D).
Molecular construction, expression, and functional characterization of IL-12 FPs
We used an IL-12 p70 fusion of p40 and p35 we had made and characterized (Skrombolas and others 2015) to construct a panel of 6 different IL-12/scFv FPs using the Gibson assembly method that allows for the precise joining of multiple overlapping DNA fragments (Gibson and others 2009). Combinations of 4 fragments were knitted together to assemble the panel of FPs containing IL-12 with the 3 different scFv (clones 1, 7, 8) containing a MMP cleavage sequence (GPLGVRG) we have termed MMPcs1 or a scrambled control sequence (GVRLGPG) (Fig. 1). The scrambled sequence is not efficiently cleaved by MMP (Bremer and others 2001).
After sequence confirmation of the 6 different IL-12 FPs, we expressed all 6 proteins using the baculovirus system, purified them using the 6x His tag and Ni-NTA agarose, and assessed their expression by western analysis using an anti 6x His antibody. As illustrated in Fig. 3A, all 6 IL-12 FPs were successfully expressed and had the expected molecular weight for the complete IL-12/scFv FP.

Biochemical and proliferative analyses of FPs containing different scFv.
All of the scFv selected inhibit the functional activity of IL-12 when present in the FP
The phage ELISA results indicated that the scFv identified bound to different epitopes of IL-12. However, it was uncertain which, if any, of these scFv would bind in the context of the FP given the physical constraints imposed by the linker and the 3-dimensional organization of the components of the FP. An additional uncertainty was whether the scFv binding to IL-12 would actually block the functional activity of the cytokine. Furthermore, it was possible that different scFv, which bind to different epitopes and have different affinities, might differ in their ability to block the functional activity of the cytokine in the intact FP. Indeed, our initial rationale for generating a panel of scFv was that we would need to identify a scFv that would neutralize the functional activity of IL-12 by binding to a neutralizing epitope.
To test whether the IL-12 was indeed functionally inhibited, we examined all 6 of the FPs in the CTLL-2 proliferation assay. The CTLL-2 cell line is widely used to measure IL-2 functional activity (Gillis and others 1978; Baker and others 1979), but can also be used to measure functional IL-12 (Khatri and others 2007; Skrombolas and others 2015). Equal molar amounts of either free IL-12 or the individual IL-12 FPs were serially diluted and cultured with CTLL-2 cells and cellular proliferation was measured. As shown in Fig. 3B, the IL-12 FPs containing either the MMPcs1 cleavage sequence or the scrambled control sequence and each individual scFv (scFv1, scFv7, and scFv8) showed ∼20-fold inhibition in functional IL-12 measured by cellular proliferation compared with the same molar amount of free IL-12.
Figure 3C is a graphical summary of the individual experiments shown in Fig. 3B, in which the percent proliferation of each FP is compared with the amount of proliferation of an equal amount of free IL-12, which is set to 100%. All 6 FPs showed reduced cytokine activity. Surprisingly, given that these scFv bind to different epitopes, these experiments did not reveal any striking differences in their ability to inhibit IL-12 in the different FPs.
All 6 IL-12 FPs are also inhibited in their ability to induce IFN-γ compared with equal molar amounts of free IL-12
Another key functional activity of IL-12 is to induce the expression of IFN-γ. To test whether the FPs were inhibited in this functional activity we cultured BALB/c spleen cells with serial dilutions of equal molar starting amounts of either free IL-12 or the individual IL-12 FPs containing either the MMPcs1 cleavage sequence or the scrambled control sequence and the individual scFvs (scFv1, scFv7, and scFv8) for 24 h and analyzed the levels of IFN-γ in these cultures.
As shown in Fig. 4, the activity of IL-12 in the intact FPs is inhibited 30- to 65-fold compared with the free cytokine in engendering an IFN-γ response in spleen cell cultures. All of the FPs were inhibited in the cytokine secretion assay. However, the IL-12/scFv8 FP containing the scFv reactive with the p35 subunit appeared somewhat less inhibited. The 2 other FPs contained scFv that recognize p40. Since the scFv clone 1 recognized a neutralizing epitope on the p40 subunit of IL-12, we focused on this FP for further analyses.
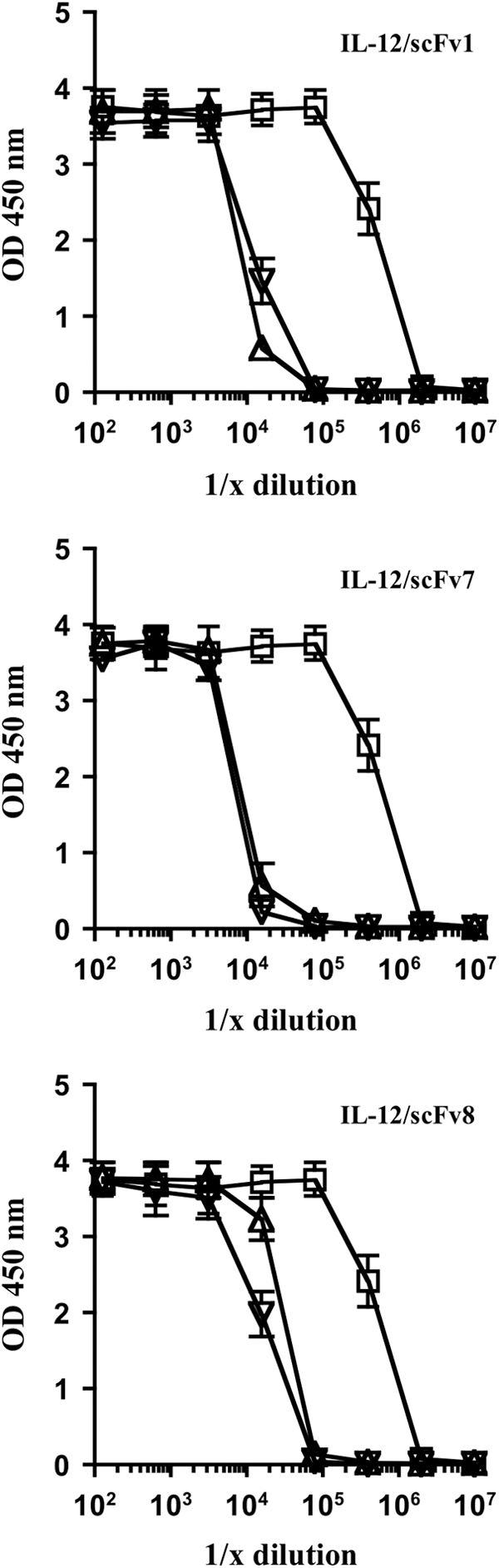
FPs ability to block IL-12 activity results in decreased expression of IFN-γ by spleen cells compared with free IL-12. BALB/c spleen cells were cultured with serial dilutions of equal molar starting amounts of either free IL-12 (open squares) or the individual IL-12 FPs containing either the MMPcs1 (open triangles) or the scrambled control sequence (open inverted triangles) for 24 h and the levels of IFN-γ in these cultures were analyzed by ELISA. The FP containing scFv1 is shown in the top graph, scFv7 in the middle graph and scFv8 in the bottom graph. The compiled data for 3 individual experiments are shown here. The degree of functional inhibition ranged from 55-fold for scFv1, 65-fold for scFv7, and 31-fold for scFv8. IFN-γ, interferon-gamma.
The IL-12/scFv1 FP containing MMPcs1 could be cleaved specifically by MMP9
We tested whether the MMPcs1 cleavage sequence was accessible and could be cleaved by MMP9. To test for cleavage, we incubated the IL-12/scFv1 FP containing the MMPcs1 cleavage sequence or the scrambled control sequence with active MMP9. The cleavage was assessed by immunoblot analysis using 2 different antibodies; an anti-IL-12primary antibody (C17.8) or an anti-penta-His antibody, which recognized the 6x His tag on the scFv portion of the FP (Fig. 1). When the FPs were treated with MMP9, the IL-12/scFv1 FP containing the MMPcs1 cleavage sequence resulted in a product of the predicted size of IL-12 detected using an anti-IL-12 antibody (Fig. 5).
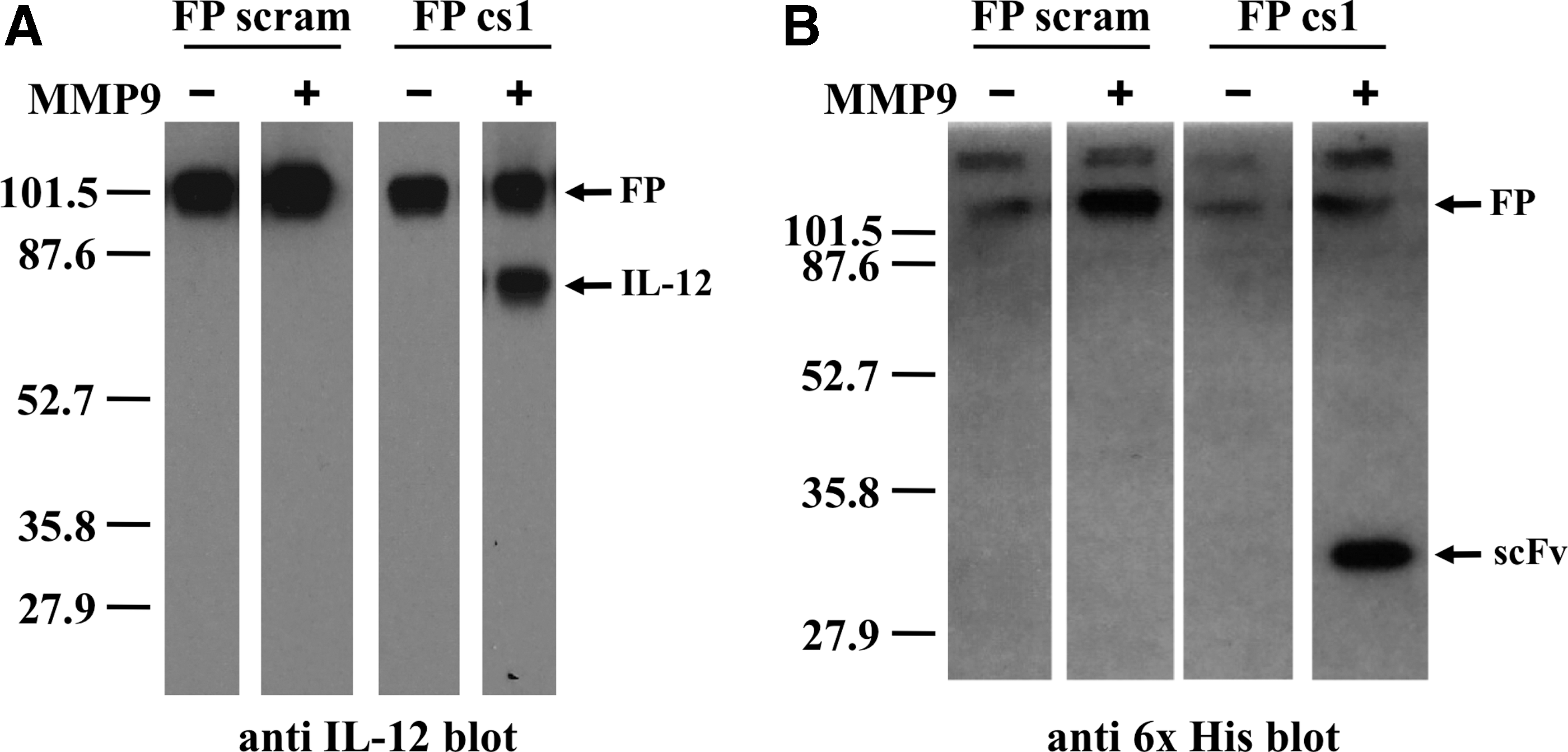
The IL-12/MMPcs1/scFv1 FP is specifically cleaved by MMP9. The IL-12/scFv1 FPs containing either the MMPcs1 or the scrambled control sequence were incubated with active MMP9 or buffer control and the resulting material tested by western analysis. Immunoblot analysis of the digested samples using an anti-IL-12 detection antibody shown on the left
Furthermore, when we assessed an aliquot of the same sample by immunoblot analysis using an antibody that recognized the 6x His tag, we observed a band around the predicted size of the 6x His-tagged scFv product (Fig. 5). These results suggest that the cleavage sequence was accessible since the FP was cleaved by the MMP9 enzyme. Moreover, the IL-12/scFv1 FP containing the scrambled control sequence was detected only in the complete, intact form by both antibodies (Fig. 5) whether or not MMP was added. This illustrates that the scrambled sequence is not efficiently cleaved by MMP9. Taken together, these results indicate that the FP can be cleaved specifically by MMP9 releasing IL-12.
Cleavage of the IL-12/scFv1 FP by MMP9 increases the functional activity of IL-12
We next analyzed whether cleavage of the FP resulted in increased biological activity of the cytokine. We tested the ability of the FP before and after cleavage to stimulate the IL-12 responsive cell line CTLL-2. The IL-12/scFv1 FP containing the MMPcs1 cleavage sequence or the scrambled control sequence were either treated or not treated with MMP9. After incubation, the samples were compared for their ability to stimulate proliferation of the CTLL-2 cell line. Strikingly, incubation of MMP9 with the IL-12/scFv1 FP containing the MMPcs1 cleavage site exhibited dramatically increased functional activity of IL-12 in comparison to the untreated FP (Fig. 6A).
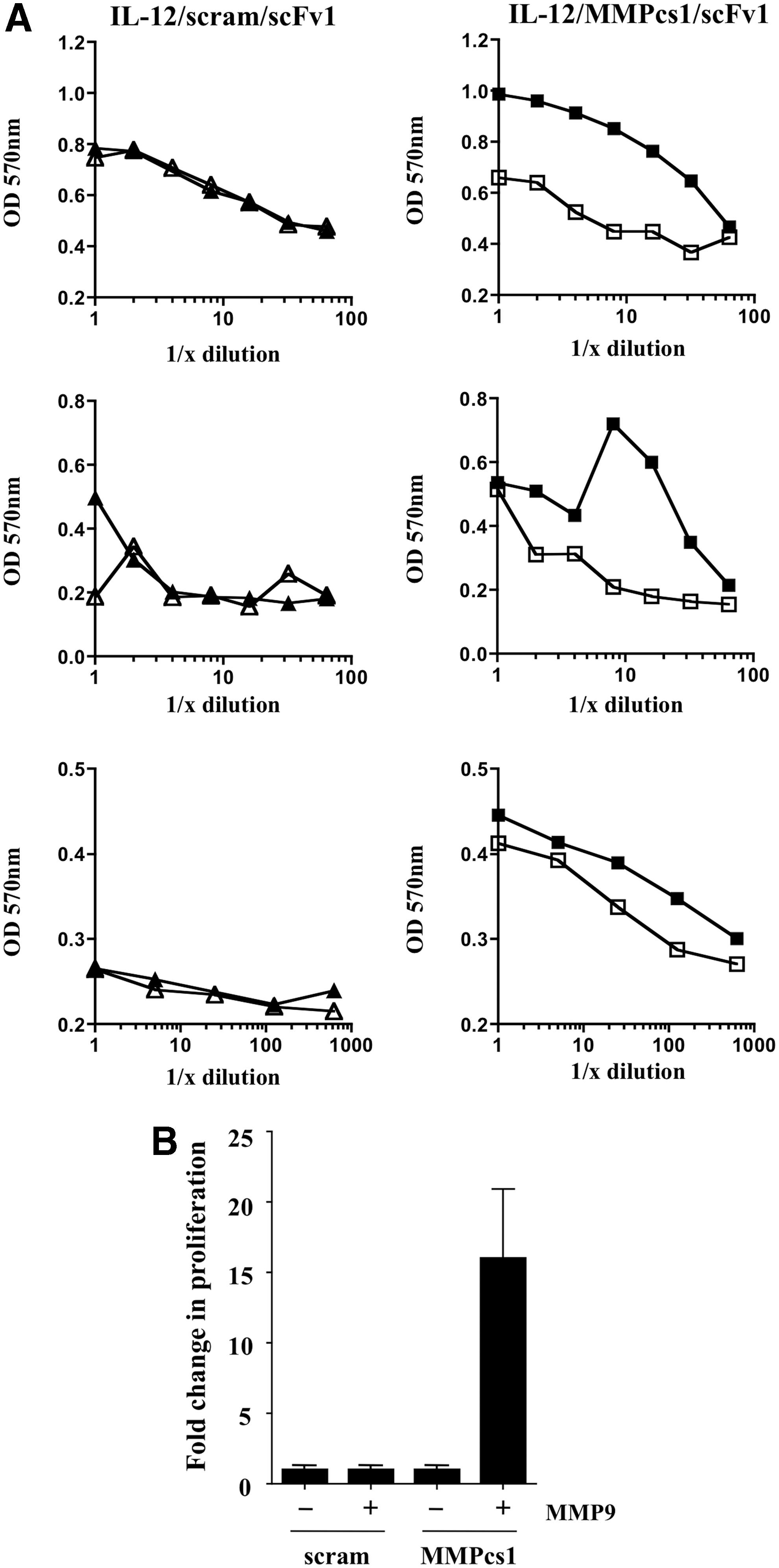
The cleavage of the IL-12/MMPcs1/scFv1 FP results in enhanced IL-12 functional activity. The IL-12/scFv1 FPs containing either the MMPcs1 or the scrambled cleavage sequence were incubated with active MMP9 or buffer control and these products were then tested in a CTLL-2 proliferation assay at various dilutions shown in
In striking contrast, the IL-12/scFv1 FP containing the scrambled control sequence did not show enhanced IL-12 activity whether or not it was incubated with MMP9 (Fig. 6A). The compiled data for 3 independent experiments (Fig. 6A) are summarized in Fig. 6B, in which the amount of proliferation seen in the absence of MMP9 was set to 1 and the increase in proliferation in the MMP9 incubated material was calculated. Taken together, these data show that the intact FP shows inhibition of IL-12, however, IL-12 biological activity increases upon protease cleavage.
Discussion
Several lines of evidence indicate that it is feasible to construct an IL-12 FP that can become active after specific protease cleavage
Several important conclusions can be drawn from these studies. First, surprisingly, all 3 of the scFv tested block the functional activity of IL-12 in the intact FPs. Second, the MMPcs1 cleavage sequence is accessible and can be cleaved by MMP9 as assessed biochemically by immunoblot analyses. Finding that the FP containing the scrambled control cleavage sequence was not cleaved by MMP9 illustrated the specificity of cleavage. Third, protease treatment of the FP (MMPcs1) resulted in dramatically increased functional activity of IL-12 illustrated by an increase in the proliferative activity of CTLL-2. Collectively, these results illustrate that the FPs are largely inactive in the intact form but can be cleaved and become functionally more active after protease treatment.
Molecular mechanisms of inhibition of IL-12 in the context of the FP
One of the most surprising findings of the current work was that all of the scFv inhibited the function of IL-12 in the context of the IL-12 FP. Before this work was begun, it was unclear if any of these scFv would block the functional activity of the cytokine. It is well appreciated that not all antibodies that bind a protein are able to inhibit their function. In the case of cytokines, the precise binding site of the antibody on the cytokine, the affinity of the antibody, as well as the relative affinity of the cognate cytokine receptor likely all play important roles. Strikingly however, despite recognizing independent epitopes, all of the scFv tested inhibited the function of the cytokine when incorporated into the FP with IL-12.
Although we have not measured the affinity of these scFv directly, it is unlikely that all of them are high affinity since they have not undergone the process of affinity maturation. While in some cases, scFv isolated from phage display can have high affinity, scFv that have not been further modified are often in the 100 nM to 1 μM range (Wuertzer and others 2008). In comparison, the dimeric IL-12 receptor is high affinity having a Kd in the pM range (Presky and others 1996, 1998).
Given the likely large difference in affinities between the scFv and the IL-12 receptor on cells, how can one account for the blocking of IL-12 in the intact FP? We hypothesize it may result from at least 2 distinct considerations. First, we propose that the binding is largely driven by proximity to the target, as the scFv and IL-12 are on the same molecule. In essence, this dramatically increases the concentration of the scFv with respect to its target, and thus it is effectively a zero-order reaction. As a result, even though the scFv can dissociate from the cytokine in the intact FP, it likely rebinds very quickly. In some sense one can consider this as a very specialized form of protein folding, in which 2 separate protein domains are “stapled” together and thus spend the vast majority of the time bound together.
After cleavage and subsequent dissociation of the scFv based on its Kd, the cytokine becomes available to bind to higher affinity receptors on cells. Thus, rebinding of the scFv to the cytokine after FP cleavage and scFv dissociation is unlikely to occur due to competition for IL-12 by higher affinity cellular receptors. Second, although we initially thought the scFv would need to bind to a neutralizing epitope, this turned out to not be the case. Rather, as the scFv binds to the cytokine, the linker, of necessity must cross the face of the cytokine. Thus, the linker, rather than being simply an inert tether as one might initially consider, likely sterically blocks interaction between IL-12 in the intact FP and receptors on target cells. This model would explain the surprising result that all the scFv inhibited IL-12 in the context of the FP.
Choice of MMP to activate the IL-12 FP
A critical feature of the FP approach and its ultimate use in vivo is its activation by proteases. While a variety of proteases have been reported to play roles in tumor development and progression (Mason and Joyce 2011), we and others are investigating using MMPs as a means of activating therapeutic molecules preferentially in the tumor microenvironment (Puskas and others 2011; Zhong and others 2013; Mullen and others 2014b). There are several reasons why MMPs are excellent candidates for activating the FPs. MMPs are a family of zinc-dependent proteases that are intimately involved and likely major contributors in tumor development and metastasis (Chambers and Matrisian 1997; Hofmann and others 2000; Lynch and Matrisian 2002; Kessenbrock and others 2010).
Because of their potent activity, MMPs are normally tightly controlled through transcriptional regulation, posttranslational mechanisms, and the coordinate expression of protease inhibitors that can block their function (Fu and others 2008; Kessenbrock and others 2010). Thus, MMP functional activity is carefully orchestrated under normal circumstances in contrast to tumors (Matrisian 1999; Egeblad and Werb 2002; Lynch and Matrisian 2002; Kessenbrock and others 2010). Interestingly, MMPs are often dramatically overexpressed at tumor sites not only due to high expression by tumor cells themselves, but in some cases also due to expression by TAMs or other myeloid cells with an M2 phenotype (Matrisian 1999).
The striking overexpression of functional activity of MMPs at tumor sites compared with normal tissues has been elegantly demonstrated in vivo using reporters that become fluorescent after protease cleavage (Bremer and others 2001; McIntyre and others 2010; Nguyen and others 2010; Xie and others 2012). Importantly, several groups have identified specific amino acid sequences recognized as substrates by various MMPs (Kridel and others 2001; Turk and others 2001; Chen and others 2002). Thus, we can exploit these key features of MMPs to localize cytokine activity to the sites of tumors with the ultimate goal of utilizing the FPs to induce robust antitumor immune responses with minimal side effects.
The protease-activated cytokine approach is fundamentally different than previous approaches exploiting proteases to activate proteins or drugs
A previously described strategy used the latency-associated protein (LAP) of transforming growth factor-beta 1 (TGF-β1) fused through a MMP cleavage site to IFN-β (Adams and others 2003; Mullen and others 2014a, 2014b). In contrast to the current work, this strategy relies on steric hindrance alone to block protein activity. As discussed above, steric hindrance may play a role in blocking cytokine function in the protease-activated cytokine approach, however, a key feature of the current work is the specific binding of the scFv that makes this approach generalizable to essentially any immunomodulator.
The protease-activated cytokine approach has some similarities to small molecule prodrugs that are cleaved to more active forms. For example, there are forms of doxorubicin that have been designed to be activated by MMP or cathepsin B proteases, thereby limiting negative side effects (Albright and others 2005; Zhong and others 2013; Anderson and Cui 2017). However, it should be noted that the prodrug application, although similar in some respects, is fundamentally different than the current work since the cytokine is not intrinsically toxic, but rather acts to amplify an immune response. The FP strategy thus could have a cascade effect by stimulating responding T cells and NK cells, which in turn can divide and produce downstream factors.
Applications of the protease-activated IL-12 FP in vivo
A major conceptual advantage of the activatable FP concept is that it is designed to be inactive until cleaved by proteases overexpressed at the tumor site. This would result in locally restricted activity of the cytokine, mimicking the natural biology of many cytokines that have evolved to act over short distances between cells. It is anticipated that this would reduce negative systemic effects while maintaining cytokine efficacy. This is particularly important for the use of IL-12 immunotherapy in patients.
The most direct application of the FP approach is to boost existing, ongoing endogenous antitumor immune responses. It is now evident that immune responses occur to many human tumors and play a key role in the success of checkpoint inhibitors, such as anti-programmed death-ligand 1 (PD-L1), anti-cytotoxic T lymphocyte-associated protein 4 (CTLA4), and anti-PD-L1 therapy that prolong and enhance T cell responses (Brahmer and others 2012; Zitvogel and Kroemer 2012; Duraiswamy and others 2013; Wolchok and others 2013; Sharma and Allison 2015). Protease-activated cytokines and checkpoint inhibitors are likely to be used effectively together since they act by different mechanisms and thus might have synergistic effects.
The protease-activated cytokine approach could also be incorporated into use with other immunotherapy strategies. For example, large-scale sequencing and bioinformatics coupled with biochemical approaches (Yadav and others 2014) have suggested it is possible to identify tumor-specific neo antigens. Therapeutic immunization with neo antigens is now being examined in clinical studies with some significant success (Carreno and others 2015; Ott and others 2017; Sahin and Tureci 2018).
Recent data support the concept of epitope spreading in cancer immunotherapy suggesting that immunization with specific tumor antigens can result in broader, more efficacious antitumor immune responses likely resulting from secondary antigen release upon tumor destruction in the context of inflammatory cytokines, such as IFN-γ (Vanderlugt and Miller 2002; Gulley and others 2017; Kueberuwa and others 2018).
Combinations of various forms of active immunization together with the protease-activated cytokine might enhance and expand such responses. Interestingly, there have also been remarkable clinical successes that have also highlighted the potential of adoptive therapy approaches, particularly those using genetically modified T cells (Chinnasamy and others 2012; Grupp and others 2013; Sadelain and others 2013). In this regard, gene therapy approaches are being used that incorporate immune modulatory genes into T cells, including cytokines such as IL-12 (Chinnasamy and others 2012; Kueberuwa and others 2018), but in some cases this results in unwanted side effects (Heemskerk and others 2008; Zhang and others 2015). Incorporating the protease-activatable IL-12 FP into adoptively transferred T cells could provide another measure of safety while maintaining efficacy.
The FP approach we have developed can be applied to other cytokines and proteases and has the potential to be a platform technology
A major unanswered question we have addressed was whether the activatable FP concept previously tested with IL-2 (Puskas and others 2011; Skrombolas and Frelinger 2014) could be applied to other cytokines. In this study, we extended this approach to a particularly potent cytokine IL-12 and showed that IL-12 function was dramatically inhibited in the intact FP but could be activated by protease treatment.
IL-12 is a particularly challenging cytokine for validating this approach since it is a heterodimeric molecule which required the initial generation of a fusion of the p40 and p35 subunits with the subsequent joining of the scFv. Initially it was unclear whether such a complex protein would fold correctly and if the scFv could even bind given the physical constraints of the linker. The data presented in this study support the idea that the domain structure of the subunits of this cytokine, and likely others, is very robust. If so, this would imply that this approach could be employed for many other cytokines and extended to other immunologically important molecules such as chemokines.
Interestingly, scFv may be ideally suited to bind and block activity of the immunomodulator since it is possible to identify scFv in vitro using phage display that bind to essentially any protein in the absence of any tolerance considerations. Importantly, given advances such as the Gibson reaction (Gibson and others 2009), alternative protease sites could be easily incorporated into these FPs and thus could ultimately be tailored to the protease profile of individual tumors. While beyond the scope of the current studies, in vivo testing of these FPs and devising efficient means of obtaining therapeutic levels of the FP will be critical in determining their potential as effective agents for cancer therapy.
Footnotes
Acknowledgments
This work was supported in part by CA184433 and PC160668 CDMRP. D.S. was supported in part by the National Institutes of Health Training Grant AI007285, and the authors want to also acknowledge generous support from Steven and Alison Krausz and from F.C. Blodgett. The authors thank Karli Norville for helpful comments to the article.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.