
Abstract
Fibrosis is a highly conserved and coordinated wound healing response to injury. In the liver, injury is promoted by immune effector mechanisms that are common across various disease etiologies and even between organs such as lungs, kidneys, heart, and other organs. Thus, the liver represents a useful model to study inflammation and repair, particularly as it is frequently biopsied in clinical contexts. Currently, strong evidence implicates IFNL3/4 polymorphisms and interferon (IFN)-λ3 levels as determinants of the extent of hepatic inflammation and fibrosis in viral and nonviral liver diseases, as well as in governing the severity of nonhepatotropic viral diseases. Interestingly, IFNL3/4 polymorphisms and IFN-λ3 levels correlate with fibrosis extent in other organs such as the lung and kidney. In this review, we discuss the association between IFN-λ and tissue inflammation and fibrosis in human disease and the potential clinical utility of the findings.
Liver Fibrosis
Chronic noncommunicable diseases characterized by tissue fibrosis account for ∼45% of deaths in Western countries (Wynn 2008). End-stage liver disease is an exemplar of this phenomenon as fibrosis culminating in cirrhosis is the cause of >90% of liver-related morbidity and mortality (Iredale 2007). Unfortunately, despite an urgent need, there are no approved drugs that specifically target tissue fibrosis, so a better understanding of its pathophysiology is critical.
In epithelial organs, including the lung, liver, skin, and kidney, in response to chronic injury, a collagen-rich scar replaces the normal tissue architecture. This process of scarring is a highly conserved and coordinated wound healing response to injury (Friedman and others 2013); conserved core features and regulatory pathways govern the process across multiple organs. Hence, a multiorgan approach was recently suggested to be useful to identify generic antifibrotic targets (Friedman and others 2013; Pellicoro and others 2014).
As a healing response, in virtually all instances, deposition of excess matrix is preceded by inflammation, with immune cells and pathways crucial in defining the phenotype (Friedman and others 2013; Pellicoro and others 2014). The latter has the capacity to exert both injury-inducing and repair-promoting effects (Pellicoro and others 2014). In this context, the liver represents a useful model for the study of inflammation and repair, particularly as it is frequently biopsied in clinical practice. Thus, findings in the liver can be informative for fibrosis of other organs.
Interferon Lambda and Liver Diseases
Since the discovery of interferon lambda (IFN-λ, 1–3) in 2003, accumulating evidence has identified their diverse contributions to antiviral and antibacterial defenses, to antitumor effects, and to autoimmunity, allergic diseases, and other immune responses (Kotenko and others 2003; Sheppard and others 2003). A fourth member, IFN-λ4, was added to the family in 2013 (Prokunina-Olsson and others 2013).
A special focus on IFN-λ in liver disease stems from the initial discovery in multiple simultaneous genome-wide association studies in 2009 of polymorphisms (rs12979860 and rs8099917) near the IFNL3/4 gene locus being the single strongest predictor of spontaneous and interferon treatment-induced hepatitis C virus (HCV) clearance (Ge and others 2009; Suppiah and others 2009; Tanaka and others 2009). This association was confirmed across different virus genotypes and in patients of different ethnic backgrounds (Asselah and others 2012; Eslam and others 2014). Subsequently, this led to a period when testing for IFNL3/4 polymorphisms was routine before treatment, was used for stratification in clinical trials, and spurred the development of PEGylated IFN-λ as a treatment. These initial discoveries also improved our understanding of the role of lambda interferons in the pathogenesis of HCV infection and more broadly in viral infections of epithelial tissues. Details of these aspects are covered in other recent reviews (Eslam and George 2015, 2016; Wack and others 2015; Hemann and others 2017).
IFNL3/IFNL4 Variants, Hepatic Inflammation, and Fibrosis
As fibrosis is largely an immune-mediated process preceded by inflammation, and HCV is noncytopathic, liver injury is mediated by the host response to the virus. The large impact of IFNL3/IFNL4 variants on hepatitis C clearance and the fact that IFN-λ is the primary interferon class in livers of humans, chimpanzees, and primary human hepatocytes (PHHs) infected with hepatitis C (Thomas and others 2012) encouraged an investigation of the role of these variants in hepatic inflammation and fibrosis.
Several reports summarized in Table 1 have now definitively demonstrated an association between the major IFNL3/IFNL4 genotypes and the severity of hepatic inflammation and consequently fibrosis. Studies have also demonstrated an association with more rapid fibrosis progression in longitudinal studies. In 2 such reports, one from the Swiss Hepatitis C Cohort Study Group (∼1,300 patients) (Bochud and others 2012) and another from the International Liver Disease Genetics Consortium (n = 4,172) (Eslam and others 2015), the IFNL3/IFNL4 major genotype was associated with more severe liver disease, particularly in those infected with HCV nongenotype 1. However, these findings have not been reported in all studies (Noureddin and others 2013). An association with liver disease severity and more rapid fibrosis progression has also been reported in patients with HCV/HIV coinfection (Guzman-Fulgencio and others 2013).
Studies of IFNL3/IFNL4 Variants and Their Association with Liver Fibrosis
Fabris and others (2011) included 2 cohorts of patients, the first cohort included patients who underwent OLT for end-stage liver disease due to different etiologies, including HCC. The second cohort included patients with chronic liver disease due to hepatitis B, hepatitis C, or alcohol abuse, a proportion had HCC.
Univariate analysis.
Association with IFNL3/IFNL4 minor genotypes.
For rs12979860 TT genotype.
CI, confidence interval; CLD, chronic liver disease; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HR, hazard ratio; LT, liver transplantation; NAFLD, nonalcoholic fatty liver disease; OLT, orthotopic liver transplantation; OR, odds ratio.
Interestingly, the association of fibrosis with the major IFNL3/IFNL4 genotype is not restricted to patients infected with HCV. Thus, similar patterns have been reported in chronic hepatitis B (Lee and others 2013; Eslam and others 2015) and even in nonviral diseases such as nonalcoholic fatty liver disease (Eslam and others 2015). The latter has been confirmed in a cohort of 946 Italian patients with biopsy-proven nonalcoholic fatty liver disease (NAFLD) (Petta and others 2017) and in another cohort of 216 patients (Uygun and others 2017). As would be expected, the association has been extended to those diagnosed by noninvasive fibrosis staging algorithms. For example, in a cohort of 266 untreated patients with HCV infection followed annually with liver stiffness assessment using Fibroscan for 5 years, the IFNL3/IFNL4 genotype was similarly associated with the risk of fibrosis progression (Boglione and others 2017). Consistently, in 304 HIV/HCV-coinfected patients, the IFNL3 rs12979860 CC genotype was associated with a higher prevalence of cirrhosis, as assessed by elastography (Barreiro and others 2011). Attesting to the robust nature of the observations, in a recent study of 116 patients transplanted for HCV infection and with available liver biopsies and donor/recipient DNA, those with IFNL4 rs368234815 TT/TT (major genotype) had a 3.45 adjusted odds of developing early post-transplant fibrosis with significantly decreased overall survival (Aiken and others 2016). In sum, IFNL3/IFNL4 polymorphisms are associated with hepatic inflammation and fibrosis across different liver disease contexts in both transplant and nontransplant settings.
IFNL3/IFNL4 Variants Associate with Clinical Outcomes
Fibrosis is the major determinant of liver-related adverse outcomes and thus the association of IFNL3/IFNL4 with outcomes is of significant interest (Vilar-Gomez and others 2018). In the HALT-C (Hepatitis C Antiviral Long-Term Treatment Against Cirrhosis) study (n = 400), those with the rs12979860 major genotype were found to be more likely to develop adverse clinical outcomes, including mortality, liver decompensation, and hepatocellular carcinoma, compared with those with the minor genotype during 3.8 years of follow-up (Noureddin and others 2013). A subsequent study of 140 Iranian liver allograft recipients observed that subjects with the IFNL3 rs12979860 major genotype had ∼2.6-fold higher likelihood of developing cirrhosis (Fereidooni and others 2017). Not all studies have demonstrated similar associations, including a report of 264 compensated HCV cirrhotic patients followed for 14.8 years (Bruno and others 2015). In HIV/HCV-coinfected patients, the rs12979860 major genotype had a 54% increase in mortality compared with those with the minor genotype (n = 264) (Clausen and others 2012). Similarly, in a longitudinal study of 484 subjects with HIV monoinfection, the rs12979860 major genotype was associated with increased overall mortality (Parczewski and others 2012).
IFNL3/IFNL4 Variants Associate with Disease Severity of Multiple Other Viruses
Similar to viral hepatitis, IFNL3/IFNL4 variants are reported to be associated with the severity of other viral infections. For example, independent reports have demonstrated an impact of IFN-λs and IFNL3/IFNL4 variants on human T cell leukemia type-1 virus (HTLV-1)-associated myelopathy/tropical spastic paraparesis (HAM/TSP) with an increase in inflammatory markers (de Sa and others 2016; Assone and others 2018). The Andes virus, which causes the hantavirus cardiopulmonary syndrome, is another example wherein IFNL3/IFNL4 polymorphisms are associated with the severity of disease progression (Angulo and others 2015).
IFN-λ and Other Organ Inflammation and Fibrosis
The impact of IFN-λ and IFNL3/IFNL4 on disease severity and fibrosis has now been extended to other epithelial organs such as the lung and kidney. Thus, a recent Polish hospital-based cohort of 326 adult asthmatics and 111 healthy subjects demonstrated that IFNL4 rs368234815 ΔG (minor genotype) was associated with less severe asthma in women >50 years, including better bronchodilator responses and reduced corticosteroid usage; this was, however, not replicated in a European population-based cohort (Chinnaswamy and others 2017). IFNL3/IFNL4 variants and circulating IFN-λ3 have also been reported to be associated with disease activity and outcomes in chronic obstructive pulmonary disease (COPD). Thus, in 528 patients with a median follow-up of 24 months, the major IFNL3/IFNL4 genotype (rs12979860) was associated with poorer prognosis, as indicated by higher BODE scores (body–mass index, degree of airflow obstruction, dyspnea, and exercise capacity measured by the 6-min walk test), predictive scores of higher risk of mortality (Celli and others 2004). Serum IFN-λ3 protein was correlated with procollagen 3, a biomarker of collagen synthesis. While those with COPD had lower serum IFN-λ3 compared with healthy controls, patients at exacerbation tended to have higher IFN-λ3 than those with stable disease (Egli and others 2018).
IFN-α is considered pivotal in the pathogenesis of systemic lupus erythematosus (SLE) and particularly for lupus nephritis. Recently, several studies have demonstrated strong evidence for a crucial role for IFN-λ and in particular IFN-λ3 in the pathogenesis of these conditions. In a Swedish study, 65 patients with lupus nephritis and dual renal biopsies at baseline and following immunosuppressive therapy were compared with 163 controls (Zickert and others 2016). Although serum levels of both IFN-α and IFN-λ were higher in patients compared with controls, the former decreased after treatment, but IFN-λ expression remained unchanged. IFN-λ levels were higher in patients with a poor histological response to immunosuppressive therapy and decreased only in patients with a histological response. Renal tissue demonstrated expression of IFN-λ, especially in inflammatory infiltrates. Since no correlation was observed between IFN-α and IFN-λ, the authors surmised that they may regulate different aspects of lupus nephritis (Zickert and others 2016). These findings were confirmed in another study, including 93 SLE patients and 67 healthy individuals. In that study, serum IFN-λ3, but not IFN-λ1 or IFN-λ2, was significantly higher in patients compared with controls (Amezcua-Guerra and others 2017). Notably, serum IFN-λ3 correlated with the severity of lupus activity, particularly with active serosal and cutaneous disease (Amezcua-Guerra and others 2017). Another study of 261 patients with SLE and the same number of healthy controls demonstrated that both IFN-λ1 and IFN-α levels were significantly higher in lupus compared with controls and the levels were not correlated with each other. Higher levels of IFN-λ1 correlated with more tissue damage and disease activity (Oke and others 2017). Consistently, a larger recent Taiwanese study has added further genetic evidence (Chen and others 2018). In a report of 2 independent cohorts comprising a total of 1,013 patients with lupus and 1,620 controls, the IFNL3/IFNL4 variant major alleles were associated with an increased risk for lupus nephritis, while serum levels of IFN-λ3 correlated with higher disease activity and more complement depression (Chen and others 2018).
Translational Aspects
One of the main challenges following a genetic discovery is the bridge from statistical associations to clinical and translational utility (Eslam and George 2015, 2016). Indeed, recent reviews emphasize the importance of downstream functional characterization to help guide translational efforts (Eslam and George 2015; Visscher and others 2017). In this regard, the discovery of IFNL3/IFNL4 variants is perhaps one of the best examples of rapid translation to clinical practice and for the understanding of disease pathogenesis as discussed below.
Utility for Clinical Staging and Prognosis
Following the discovery of IFNL3/IFNL4 variants and their role in hepatic inflammation, work using data mining approaches demonstrated their utility in prognostic algorithms. Thus, in 2 large cohorts totaling 4,277 patients with chronic liver disease, an IFNL3/IFNL4-based decision tree was constructed that included other simple clinical factors. The decision tree had a negative predictive value of >0.96 for exclusion of cirrhosis in patients with chronic hepatitis B, chronic hepatitis C, and in those with NAFLD (Eslam and others 2016). Likewise, another IFNL3/IFNL4-based model has been developed for 427 Egyptian patients infected with chronic hepatitis C, with an overall diagnostic accuracy of 0.88 (Shousha and others 2018). In HIV/HCV-coinfected patients (n = 679), diagnostic accuracy of 0.756 was observed for prediction of 3-year risk of developing significant liver fibrosis (Moqueet and others 2017).
Can IFNL Findings Be Exploited for Therapeutic Benefit?
The restricted expression of IFN-λ compared with other types of interferons renders it a potentially attractive target for immune-based therapies of chronic inflammatory diseases (Eslam and George 2016). However, any attempt to do this first requires a better understanding of the functional mechanisms underlying the genetic association and identifying the likely causative protein for the effect. Such a systematic approach can also lead to the discovery of novel negative regulators of IFN-λ signaling that could serve as therapeutic targets.
The rs368234815 single-nucleotide polymorphism (SNP) controls the generation of a functional IFN-λ4 protein and is in high linkage disequilibrium with the rs12979860 SNP and another SNP (rs4803217) in the 3′ untranslated region of IFNL3 (Prokunina-Olsson and others 2013). The weight of current data suggests that these SNPs control IFN-λ3 expression and stability not only in the liver, serum, peripheral blood mononuclear cells, and dendritic cells from HCV-infected patients (Fukuhara and others 2010; Shi and others 2012; Yoshio and others 2013; McFarland and others 2014; Murata and others 2014) but also following infection with other viruses, such as hepatitis B virus (HBV) (Li and others 2011), cytomegalovirus (CMV) (Egli and others 2014a), and influenza (Egli and others 2014b). As expected, some reports have not confirmed the association (Ge and others 2009; Urban and others 2010). A recent study suggested that use of the droplet digital polymerase chain reaction (ddPCR) results in significantly improved performance for quantification of nucleic acid targets compared with real-time PCR and that it translates to superior diagnostic performance (Hindson and others 2013). Using ddPCR, correlation between rs12979860 and hepatic IFN-λ3 expression was recently confirmed (Eslam and others 2017).
The very high linkage disequilibrium between genetic variants in the IFNL3/IFNL4 region (Suppiah and others 2009; Tanaka and others 2009) renders it hard to dissect whether IFN-λ3 or IFN-λ4 is the more likely causative protein underlying the genetic association. In this regard, the extremely low expression of IFN-λ4 and the fact that it has not been robustly detected in livers of patients with non-HCV liver diseases or in immune cells suggest that the association with inflammation and fibrosis is likely related to IFN-λ3 rather than IFN-λ4 (Hong and others 2016). Since the IFNL4 region is absent from rat and mouse genomes, unequivocal confirmation of the above is problematic (Key and others 2014).
An interesting study from the Hartmann laboratory recently provided an important clue. Based on coinheritance patterns of the 2 IFNL4 variants, rs368234815 and rs117648444, the authors demonstrated that variants control the generation of IFN-λ4 with different biological activity and a differential impact of the highly active IFN-λ4-Pro70 and the less active IFN-λ4-Ser70 on viral clearance (Terczynska-Dyla and others 2014). Using a similar approach, it was recently suggested that IFN-λ3, but not IFN-λ4, is likely the causative protein that governs inflammation and fibrosis since there was no differential effect of the IFN-λ4-Pro70 and IFN-λ4-Ser70 isoforms on hepatic inflammation and fibrosis or related features despite their discrepant activity (Eslam and others 2017). Consistently, a recent elegant report provides mechanistic evidence that humans suppress IFN-λ4 expression (Hong and others 2016) and that IFN-λ3 is induced several log-fold higher than any of the IFN-λ4 splice forms (Hong and others 2016). This observation was confirmed in another study (Bamford and others 2018). Based on their findings, the authors suggest that the contribution of IFN-λ4 in physiological immune contexts is relatively low and thus likely dependent on other IFN-λ family members (Hong and others 2016). Notably, multiple lines of evidence suggest that IFN-λ4 behaves like a classic IFN-λ and induces a similar set of interferon-stimulated genes as IFN-λ3 (Hamming and others 2013; Lauber and others 2015; Onabajo and others 2015), rendering the possibility that IFN-λ4 can have a protective anti-inflammatory effect unlikely. In this respect, neither secreted nor intracellular IFN-λ4 isoforms interfere with type I or III IFN signaling (Hong and others 2016).
Although the exact mechanisms of how IFN-λ, particularly IFN-λ3, is involved in inflammation and fibrosis are incompletely understood, our knowledge is evolving and is summarized in Fig. 1. Serum IFN-λ3 levels correlate with inflammatory and fibrogenic markers (Aoki and others 2015), and subjects with the major IFNL3/IFNL4 genotype (IFN-λ3 producers) have higher hepatic and plasma inflammatory markers (Holmes and others 2015). Infiltration of immune cells to the liver plays a pivotal role in the progression of disease (Wynn and Barron 2010). Consistently, subjects with the major IFNL3/IFNL4 genotype have lower blood immune cell populations, particularly monocytes, suggesting their migration to the liver or other sites of inflammation (O'Connor and others 2016). The latter has been confirmed by reports demonstrating increased immune cell infiltration in the liver of patients with the major genotype (Honda and others 2014; Eslam and others 2017). Macrophages are considered master regulators of hepatic inflammation and fibrosis, and sCD163, a macrophage activation marker, has been shown to strongly correlate with liver fibrosis (Wynn and Barron 2010; Kazankov and others 2016). Interestingly, carriers of the major IFNL3/IFNL4 genotype have higher serum sCD163 compared with those with the minor genotype (Shirasaki and others 2014; Eslam and others 2017). Furthermore, IFN-λ3 drives macrophages toward a proinflammatory phenotype and increases the chemotaxis of other immune cells, including T cells (Read and others 2018). Metallothionein expression was also reported to be increased in those with the major genotype and correlated with hepatic fibrosis (O'Connor and others 2014). Finally, a recent study that used laser capture microdissection of HCV-infected and adjacent PHHs has shown that the principal activated pathways in cells from donors with the major IFNL3/IFNL4 genotype (IFN-λ3 producers) are cell death and survival, while they were antimicrobial in those with the minor genotype (Sheahan and others 2014).
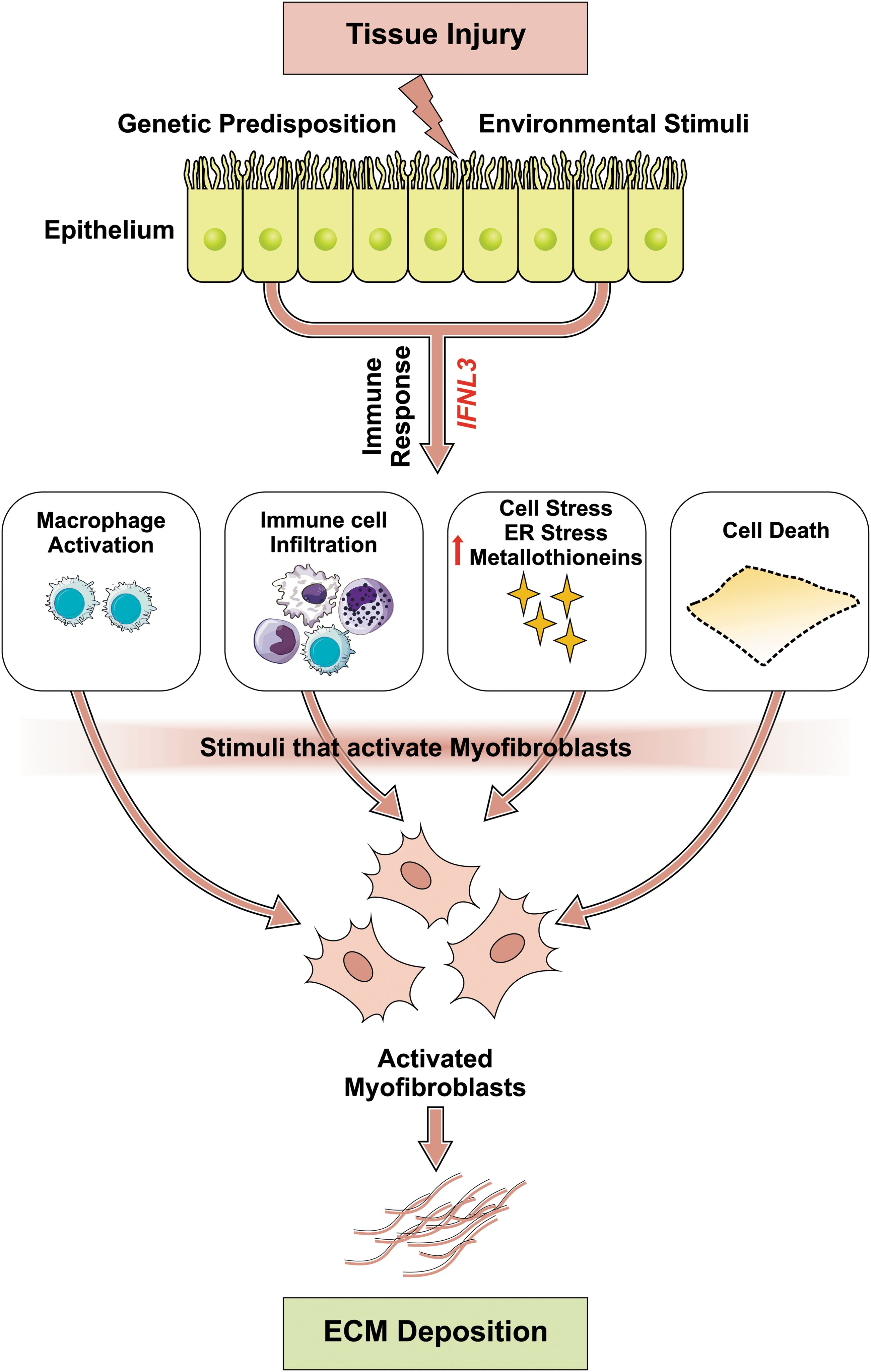
Mechanisms by which IFN-λ3 can lead to organ fibrosis. A range of injurious stimuli initiate activation of distinct immune cell subsets and IFN-λ3 generation in epithelial cells. IFN-λ3 induces several distinct profibrotic programs in epithelial cells. It can skew cells toward cell death, which is reported to initiate fibrosis in various models of liver fibrosis. IFN-λ3 leads to increases in metallothionein expression, ER stress, and production of ROS. IFN-λ3 promotes recruitment and activation of a variety of immune cell subsets in particular macrophages, which, in turn, release fibrosis-promoting cytokines. These, in turn, enhance further recruitment of immune cells and secretion of IFN-λ3. These events lead to expansion, activation, and/or recruitment of tissue myofibroblasts, the primary source of pathologic ECM that characterizes tissue fibrosis in organs such as the liver, kidney, and lung. ECM, extracellular matrix; ER, endoplasmic reticulum; IFN-λ, interferon lambda; ROS, reactive oxygen species.
Finding novel, specific negative regulators of IFN-λ3 signaling to dampen inflammation is a challenge. In this regard, a recent study from our laboratory demonstrated that zinc is a specific negative regulator of IFN-λ3 (Read and others 2017). Whether zinc can be used as a therapeutic strategy to dampen IFN-λ3-mediated chronic liver injury will, however, need further investigation.
In conclusion, IFNL3/IFNL4 variants and IFN-λ3 are associated with hepatic inflammation and fibrosis across different etiologies, as well as in organs such as the lung and kidney. Based on current evidence, IFN-λ3 rather than IFN-λ4 likely mediates the inflammatory effect through mechanisms, including promoting immune cell migration to the liver, activating macrophages, promoting cell death, and increasing the expression of metallothioneins. From a translational perspective, IFNL3/IFNL4 variants can be incorporated into diagnostic and prognostic algorithms for staging liver fibrosis. While zinc appears to be a novel and specific inhibitor of IFN-λ3 activity, mechanistic studies to identify other negative regulators of IFN-λ3 activity and clinical trials of the efficacy of zinc treatment to downregulate chronic tissue inflammation would be worthwhile.
Footnotes
Acknowledgments
M.E., G.A., and J.G. are supported by the Robert W. Storr Bequest to the Sydney Medical Foundation, University of Sydney; a National Health and Medical Research Council of Australia (NHMRC) Program Grant (APP1053206, APP1149976) and Project Grants (APP1107178 and APP1108422).
Authors' Contributions
The authors contributed equally to all aspects of this article.
Author Disclosure Statement
The authors declare no competing financial interests.